

《1.引言》
1.引言
人类活动造成的温室气体排放尤其是二氧化碳(CO2)的排放近年来不断地增加。CO2加氢过程被认为是一种可行的将二氧化碳转化为有价值的化学品或燃料的方法。其加氢过程被认为主要是由两步连锁反应组成:①逆水煤气转换(RWGS)反应;②费托合成(FTS)过程[1,2]。

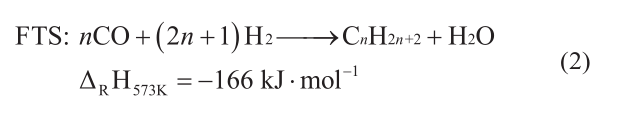
在RWGS反应中,H2与CO2反应生成水和CO,工业催化剂主要选取四氧化三铁(Fe3O4)。生成的CO作为FTS过程的气体反应物,在H2氛围下通过表面加氢-聚合反应生成碳氢化合物。FTS过程主要通过钴、铁、钌等金属催化。铁基催化剂由于具有获得容易、具有高RWGS反应活性、产生较多烯烃和支链碳氢化合物的特点,被广泛地用于CO2加氢过程[3–5]。钴基催化剂不适用于CO2加氢过程,即使在RWGS反应中加入Mn和K助剂,其转化的CO的分压仍然很低。
Riedel等[6]发现对于FTS和RWGS,在Fe催化剂中加入K助剂之后,催化活性和选择性的变化趋势相似。而RWGS过程的不同的动力学区间要比FTS过程的动力学区间要长。选择性结果显示CO2加氢反应与CO的费托过程是相伴发生的[7,8]。他们也报道了在RWGS反应生成CO的过程中,碳化铁在铁基催化表面生成[6]。对于铁基催化剂,表面碳化是展现出费托活性的前提[4,9–15]。K助剂被认为是通过提高催化剂碱性来抑制H2解离吸附作用[6,7,16,17]。通过这样的方式,它提高了烯烃的选择性,同时增加了链增长的概率,抑制了甲烷生成,促进了碳化铁的生成[15,18,19]。然而,铁表面的积碳作用会引起催化剂的失活。Mn助剂作为结构型和电子型助剂,增加了氧化铁在表面的分散程度、降低了合成气还原过程中的碳在催化剂表面的沉积作用。因此,其减小了铁基催化剂在费托合成反应中的失活速率[20–22]。Davis[23]提出甲酸中的含氧结构可能来源于链引发阶段的CO或CO2。在反应条件下,催化剂由Fe3O4组成,其表面覆盖了一层碳化铁,在反应过程中碳化铁结构一直被保留。
碳纳米管(CNTs)作为一种具有前景的催化剂载体[24–27],具有较大的表面积,可以很好地分散具有催化活性的纳米粒子[28]。其也可以防止烧结现象,因此在提高铁基费托过程催化剂的稳定性和选择性方面具有良好作用[8,17]。碳纳米管中的含氧和含氮的功能基团同时也可以作为金属活性物种的配位点[24,25]。掺氮碳纳米管(NCNTs)可以通过碳纳米管在氨气中的后处理部分氧化制备得到[24,29]。Kundu等[29]发现用氨气在673K条件下处理后的NCNTs主要含有吡啶型基团,其被认为是活性金属物种的配位点。Chew等[2]发现753K热处理5h后得到49%的FeO和51%的Fe组成的铁金属混合物。Fe/NCNT、Fe/OCNT、Fe/SiO2用于CO2加氢过程时,Fe/NCNT比Fe/OCNT具有较低的CO选择性。Fe/SiO2与碳纳米管做支撑材料的催化剂相比,在CO2加氢过程中不具备优势。基于CO2加氢反应步骤,在RWGS生成的CO通过加氢作用生成碳氢化合物。Chew等[2]发现CO在Fe/NCNT表面的氢化作用比在Fe/OCNT表面的氢化作用要快,而且在催化反应结束后,其表面均含有碳化层。
NH3分解反应是一种可以被许多过渡金属表面催化的反应[30,31]。化学吸附原子氮的重组解吸作用是NH3分解过程中的速率决定步骤[30]。因此,该方法可以用于表征添加的助剂对催化剂的热稳定性的影响。
本文关注了K和Mn助剂对于掺氮碳纳米管的铁基催化剂活性、产物选择性和热力学稳定性的影响。在NCNTs上的铁基纳米颗粒通过浸渍方法合成。催化剂的相分析通过X射线衍射(XRD)进行。H2程序升温还原(H2-TPR)和原位X射线吸收近边结构分析(XANES)被用于研究铁催化剂的可还原性。在K/Mn助剂处理的掺氮碳纳米管下的铁纳米颗粒催化剂的NH3分解和CO2加氢被用于评价催化剂性能和研究助剂的影响。近期报道的研究主要关注了助剂对产物分布的影响[32]。
《2.实验方法》
2.实验方法
《2.1.催化剂制备》
2.1.催化剂制备
本文使用柠檬酸铁胺作为铁基催化剂的前驱体,通过浸渍的方式负载于氮掺杂的碳纳米管之上。随后,通过将其在碳酸钾和硝酸锰溶液中浸渍的方法得到含有K和Mn助催化剂的铁基催化剂(K/Fe/NCNT),具体过程详见参考文献[32]。
《2.2.表征方法》
2.2.表征方法
催化组分可通过原子吸收分光光度法(AAS)进行定量分析。XRD方法用于分析催化剂中结晶相。测试设备是荷兰帕纳科公司的多功能粉末衍射仪,同时使用Cu Kα radiation(λ=1.54Å),操作条件为电流40mA和加速电压45kV。光谱扫描步长为0.026°,扫描范围2θ为20°~80°。结晶相的确定通过使用无机晶体结构数据库(ICSD)确定。H2-TPR的操作过程为:放入40mg催化剂,加热升温速率为10K•min–1,同时保持保护气Ar中含有4.73%的H2,总流量为84.1cm3•min-1。加热温度从323K到1073K,持续1h。
催化剂的还原性通过在H2氛围下的原位XANES表征。时间分辨系统为XAS beamline(BL2.2)[Synchrotron Light Research Institute(SLRI),Thailand]。能量色散单色仪中的弯曲晶面Si(111)用于对焦多色的X射线束到样品上[35]。X射线穿过样品然后分叉分别到达灵敏探测器(NMOS-linear image sensor),操作条件:采样时间为250ms。4.5mg样品被制备成直径4mm的圆片。在催化还原过程中使用Fe K-edge原位XANES。操作条件为4cm3•min-1的H2流速和80.1cm3•min-1的Ar流速。从323K以10K•min-1的加热速率加热2h至923K。铁箔用于矫正Fe K-edge在7112 eV下的吸收峰。线性回归分析用Athena实现[36]。铁箔、FeO、Fe3O4和Fe2O3被作为基准进行参考。
《2.3.催化实验方法》
2.3.催化实验方法
NH3分解作为测试实验用于分析助剂的作用和催化剂的热稳定性。NH3分解实验在U型石英反应器中进行。10mg的催化剂和100mg的SiC的混合物填充于石英棉塞之间。在催化实验前,催化剂在25cm3•min–1的He气、室温条件下净化30min。之后还原实验在流量为25cm3•min–1的H条件下,开始于673K以5K•min–1的升温速率加热1h。反应结束后,反应器在323K、流量为25cm3•min–1的He气中冷却,吹扫体系中的H。随后,反应气体(5cm3•min–1的含有10%NH3的He、He的流速为45cm3•min–1)通入反应器约20min后达到稳态。之后,反应器同样以5K•min–1的加热速率加热至923K,随后在923K的温度下加热1h。随后,反应器从923K以5K•min–1的降温速率冷却至室温。通过将反应后的气体通入非色散红外探测器(BINOS)来检测氨气浓度。
对于CO2加氢过程,40mg的催化剂和160mg的SiC的混合物填充于石英棉塞之间。SiC作为稀释剂减小放热反应过程中产生的热量。在催化实验之前,催化剂在653K和25bar压力下,维持50cm3•min–1的H2氛围中还原5h。之后,反应器通过633K的Ar以30cm3•min–1的流量冷却,吹扫反应器中的H2。反应气体(H2流速为22.5cm3•min–1、CO流速为7.5cm3•min–1、Ar流速为3.3cm3•min–1)通过催化剂床层维持空速为50L•(g•h)–1、温度为633K、压力为25bar。产物直接通过热导检测器(TCD)连接在线的气相色谱仪(GC)进行分析,其中,双火焰离子化检测器(FID)使用Ar作为内部标准。具体的实验操作过程在文献[37]中进行了详细的描述。
《3.结果与讨论》
3.结果与讨论
通过AAS分析的焙烧后的金属质量分数结果如表1所示[32]。未掺入助剂的Fe/NCNT的XRD图谱和掺入助剂的Fe/NCNT焙烧后的图谱显示,四氧化三铁(Fe3O4)是铁催化剂中除了六角形石墨的主要相[32]。
焙烧后的Fe/NCNT、K/Fe/NCNT、Mn/Fe/NCNT和K/Mn/Fe/NCNT的程序升温还原(TPR)结果如图1所示。对于助剂未处理的掺氮碳纳米管的铁基氧化物纳米颗粒[图1(a)],其在584K和613K的两个较小的峰被观测到,这分别是因为氧化铁表面物质发生还原且Fe2O3还原为Fe3O4[2]。773K处的峰由于Fe3O4进一步还原成金属铁而减少。根据式(3)和TPR结果,纳米铁氧化物逐步被还原[5,38]:
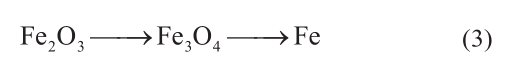
K/Fe/NCNT的TPR结果朝着高温方向(598K、636K和792K)[图1(b)]漂移表明K助剂使得催化剂还原过程被延缓[30]。与未进行助剂处理或者K助剂处理的铁基催化剂相比,Mn助剂的铁基催化剂(Mn/Fe/NCNT和K/Mn/Fe/NCNT)的开始还原温度从485K向较低温度420K漂移[图1(c)、(d)],同时也没有表面铁氧化而出现较小的峰。这一发现说明铁氧化纳米颗粒在Mn助剂处理后氧化铁颗粒更加分散,从而样品更容易被还原,这一实验结果与Tao等[20]的结果一致。对于Mn/Fe/NCNT催化剂,峰出现在较低温度(615K),此时Fe2O3被还原成为Fe3O4,同时在较高温度(753K和821K)下出现双峰是由于Fe3O4进一步被还原成为Fe[图1(c)]。还原过程中出现双峰的原因可以归因于Fe3O4被还原成FeO(作为中间产物),然后被进一步还原成Fe,而不是Fe3O4在773K的温度下被直接一步还原成Fe。Mn助剂的处理使得Mn2+进入到FeO晶格中起到稳定FeO晶格的作用[20]。与Mn/Fe/NCNT相同的是,在K/Mn/Fe/NCNT还原实验中在高温(744K和807K)下仍然发现了两个独立的峰[图1(d)],且其Fe3O4→FeO→Fe的还原温度更低,说明K助剂的加入使得FeO的稳定性降低。Pernicone等[39]指出,FeO更加容易被还原,与传统的Fe3O4催化剂相比,其对于氨合成过程具有更高的活性。因此,在Mn/Fe/NCNT中,FeO作为中间产物存在于还原过程中,进一步生产金属Fe纳米颗粒。当Fe/NCNT同时加入K助剂和Mn助剂时,FeO中间体更加被容易还原成为铁纳米颗粒。
《表1》
表1 催化剂中的金属成分[32]

《图1》

图1.H2TPR焙烧后、反应前的结果:(a)Fe/NCNT;(b)K/Fe/NCNT;(c)Mn/Fe/NCNT;(d)K/Mn/Fe/NCNT。
原位TPR XANES实验用于进一步揭示在CO2加氢过程前的H2还原过程中,铁基催化剂(如Fe/NCNT、K/Fe/NCNT、Mn/Fe/NCNT、K/Mn/Fe/NCNT)的结构演变。图2展示了在H2氛围下从323K加热到923K的4种催化剂的Fe K-edge XANES的堆叠光谱。光谱是随着温度的上升而逐渐形成的。XANES的特征变化也揭示物相变化的几个步骤,如吸收峰边缘向更低的能量方向转移的同时白线逐步消失,说明有金属铁在逐步生成,因为其光谱图逐渐变得与单质铁相似。如图3所示,线性拟合(LCF)随后被用于分析在铁基催化剂还原过程中的相成分的演化。在所有的测试组中,XANES光谱变化显示了相转化过程存在的三个步骤:Fe2O3→Fe3O4→FeO→Fe。不同的只是每种催化剂所带来的不同的反应速率和相转化的比例。这样的实验结果与我们之前未加入助剂情况下的铁基催化剂的结果相一致[2]。在反应刚开始阶段,除了K/Fe/NCNT主要是由Fe2O3组成,焙烧过后的前驱体大部分是由Fe2O3和Fe3O4组成。Fe2O3还原成Fe3O4的过程,对于Fe/NCNT、K/Fe/NCNT、Mn/Fe/NCNT和K/Mn/Fe/NCNT不同种催化剂分别开始于473K、473K、613K、523K。直到Fe2O3全部转化为Fe3O4时,相转化过程才逐渐开始,其中,K/Fe/NCNT和K/Mn/Fe/NCNT的转化温度在593K和623K。与此不同的是,对于Fe/NCNT和Mn/Fe/NCNT催化剂,Fe2O3形成的还原物中Fe3O4和氧化铁可以共存,其转化温度分别在513K和773K。Fe3O4在生成金属铁的过程中,先逐步转化为氧化铁,之后再形成金属铁。需要说明的是,在Mn助剂处理过的Mn/Fe/NCNT和K/Mn/Fe/NCNT催化剂中,Fe3O4几乎全部转化成氧化铁(大约88%~100%的氧化铁),然后再转化为金属铁单质。这一现象在其他的催化剂实验组中并未明显出现。在Fe/NCNT、K/Fe/NCNT、Mn/Fe/NCNT和K/Mn/Fe/NCNT4种催化剂中金属铁单质分别在773K、773K、823K、803K温度下生成。在923K加热2h后,每种催化剂均由金属铁单质组成,相伴有少量的氧化铁存在。
《图2》

图2.XANES光谱图。(a)Fe/NCNT;(b)K/Fe/NCNT;(c)Mn/Fe/NCNT;(d)K/Mn/Fe/NCNT。操作条件:在流量为4cm3•min–1的H2和80.1cm3•min-1的Ar的气体氛围下,从323K加热至923K,升温速率为10K•min−1。
《图3》

图3.铁基催化剂的相转化的原位XANES还原结果。操作条件:4cm3•min–1的H2和80.1cm3•min–1的Ar的气体氛围下,从323K加热至923K,升温速率为10K•min–1,在最终温度停留2h.(a)Fe/NCNT:(b)K/Fe/KNCNT:(c)Mn/KFe/NCNT:(d)K/Mn/Fe/NCNT
NH3分解实验用于分析助剂对于催化剂热稳定的影响。图4显示了在用不同方式处理的催化剂下NH3转化率随着温度变化的曲线。在空白对照实验中,NH3直到923K才开始分解,说明在923K以下NH3并不发生热分解。对于每种催化剂相同的是,NH3分解的起始温度几乎都在530K左右。Fe/NCNT催化剂组的NH3转化率在加热过程中一直增加,在923K温度下转化率可以达到98%,说明在掺氮碳纳米管下的铁基纳米颗粒对于NH3分解具有活性。当继续在923K温度下加热1h,NH3转化率从98%下降至87%,说明催化剂在逐渐失活。NH3在金属铁表面的空位处吸附之后,发生NH3分解过程[40]:

式中,*为金属Fe表面的一个空位。
《图4》

图4.不同方式处理的催化剂下NH3转化率随温度变化的曲线。
对于K/Fe/NCNT催化剂,NH3分解的起始温度为583K,与Fe/NCNT的起始温度相比来说相对较低。有文献表明,吸附的氮原子组合、脱附[式(7)]是NH3分解的决速步[30]。K助剂使得N—*更加不稳定,因此提高了K/Fe/NCNT催化剂的NH3分解的活性。除此之外,Jedynak等[41]发现K助剂处理的催化剂具有更高的铁基催化剂的分散度。但是NH3的转化率在840K左右开始下降,这是由于Fe纳米颗粒烧结所造成的。当在923K加热1h时,由于K/Fe/NCNT存在明显的钝化作用,NH3分解的转化率降至70%。
与Fe/NCNT和K/Fe/NCNT催化剂不同的是,NH3转化率在Mn/Fe/NCNT催化剂作用下达到最高的水平,几乎在99%水平维持稳定。说明在温度维持在923K时,Mn稳定了金属Fe纳米粒子。K/Mn/Fe/NCNT催化剂对于NH3分解反应展现出最优的性能,当温度达到760K时转化率达到最大值并在该温度下具有良好的稳定性。这样的结果说明K助剂作为电子助剂,使得N—*更加不稳定,因此提高了NH3转化的活性,而Mn助剂作为结构性助剂,有效阻止了Fe催化剂的严重的结焦问题。
催化剂在降温过程中的NH3分解转化率与加热过程相比较低,这是因为催化剂活性在923K下恶化。这样的现象在4种催化剂中均出现。对比降温过程的4种催化剂,K/Mn/Fe/NCNT仍然显示出最高的转化率。催化剂在降温过程中的催化活性为:在860K下,K/Mn/Fe/NCNT仍然维持在99%的转化率;Mn/Fe/NCNT转化率为90%;K/Fe/NCNT转化率为45%;Fe/NCNT转化率为65%。显然,Mn助剂加强了铁金属纳米颗粒的分散性,同时有效阻止了结焦过程[42]。
所用催化剂被用于在633K和25bar下的CO2加氢过程,反应时间为60h。在反应结束之后,催化剂通过XRD表征,如图5所示。对于Fe/NCNT催化剂[图5(a)],衍射峰出现在40.9°、43.5°和44.2°位置,表明存在χ-Fe5C2。而在30.4°和35.8°的衍射峰说明存在Fe3O4。在CO2加氢过程中,被还原的铁相之后被转化为碳化铁,其对费托反应过程具有催化活性,然而FTS过程产生的水会使部分Fe氧化生成Fe3O4。另外,即使是在60h的反应过后,K/Fe/NCNT催化剂[图5(b)]仍然保持了χ-Fe5C2为其主体相。Mn/Fe/NCNT的衍射图谱[图5(c)]表明,衍射峰出现在40.9°、43.5°和44.2°的位置,显示物质仍然为χ-Fe5C2,除此之外还存在24.3°、31.4°、51.8°位置的MnCO3的衍射峰。K/Mn/Fe/NCNT催化剂[图5(d)]中的MnCO3衍射峰也明显存在。Grzybek等[43]指出,Mn/Fe氧化物催化剂的表面存在着大量Mn元素,其大量的附着阻止了Fe元素氧化生成Fe3O4[43]。Mn元素仍然可以延缓在反应过程中氧化铁类型的纳米颗粒的聚集现象[42]。在60h反应之后,K/Mn/Fe/NCNT催化剂通过XRD图谱表征,仍然显示催化剂表面具有大量的反应活性组分χ-Fe5C2,没有可检测到的Fe3O4的特征峰。
《图5》

图5.在633K和25bar下操作60h后CO2加氢XRD图谱。(a)Fe/NCNT;(b)K/Fe/NCNT;(c)Mn/Fe/NCNT;(d)K/Mn/Fe/NCNT。
图6显示了不同的催化剂Fe/NCNT、K/Fe/NCNT、Mn/Fe/NCNT和K/Mn/Fe/NCNT对于CO2转化率随着运行时间的影响,操作温度为633K。Fe/NCNT催化的反应在最开始具有38%的CO2转化率,随着运行时间时间的增长,反应活性持续地降低。催化剂的活性降低是由于铁颗粒在高温和较长运行时间下的结焦。Mn助剂的铁基催化剂在最开始表现出较低的转化率,只有约30%,但是其催化活性的稳定性要比未加入助剂的铁基催化剂要好。Grzybek等[43]指出在反应过后催化剂表面具有大量的Mn元素。说明在反应过程中,催化剂表面大量的Mn元素覆盖了在反应中的Fe的活性位点[42],但是碳原子仍然能够通过扩散的方式,通过富有Mn元素的覆盖层,形成具有加氢活性的碳化铁[43]。因此Mn助剂是结构性助剂,可以稳定铁相的反应活性,如我们之前的TPR分析和氨分解实验,因此在60h的运行时间仍然具有稳定的CO2加氢的催化活性。K助剂处理的铁基催化剂均展现出高的CO2加氢转化率,其超过30%。通过双助剂处理的K/Mn/Fe/NCNT催化剂展现出相对较低的转化率,为30%,但是CO2加氢性能更为稳定。很显然,K助剂提升了CO2的加氢转化率,而Mn助剂从结构上加强了活性碳化铁纳米颗粒的稳定性。
Fe/NCNT、K/Fe/NCNT、Mn/Fe/NCNT和K/Mn/Fe/NCNT的产物选择性如图6(b)和表2所示。所有催化剂转化生成的CO的结果与Lee等的结果相同[44]。Wang等[1]也报道了K助剂有利于CO2的吸附过程,产生了更多CO2分解成CO的活性位点。有报道指出,FTS反应速率与RWGS反应速率相比相对较低[43],这抑制了甲烷生成而提高了长链碳氢化合物的选择性。Mn助剂处理的Fe/NCNT与未用Mn处理的Fe/NCNT具有相似的产物选择性。除此之外,并没有发现经Mn/Fe/NCNT处理的含氧碳氢化合物的形成。
《图6》

图6.(a)CO2转化率随着运行时间的变化关系;(b)产物选择性。
《表2》
表2 产物选择性、C2~C5范围内的烯烃选择性、支链增长概率(α)和CO2转化率(XCO2)在不同催化剂处理下运行60h后的结果

我们对K/Mn/Fe/NCNT的催化性能进行了更细致的研究,通过增加催化剂的量的方法,增加了在反应区间的停留时间。图7和表3显示了高的CO2转化率和加氢选择性、较低的CO选择性和较高的烯烃和乙醇选择性。
《图7》

图7.(a)CO2转化率随着运行时间变化的曲线;(b)不同停留时间下K/Mn/Fe/NCNT处理的CO2加氢反应下的产物选择性。
《表3》
表3 60h运行时间后的产物分布:(a)40mg、(b)160mg、(c)640mg
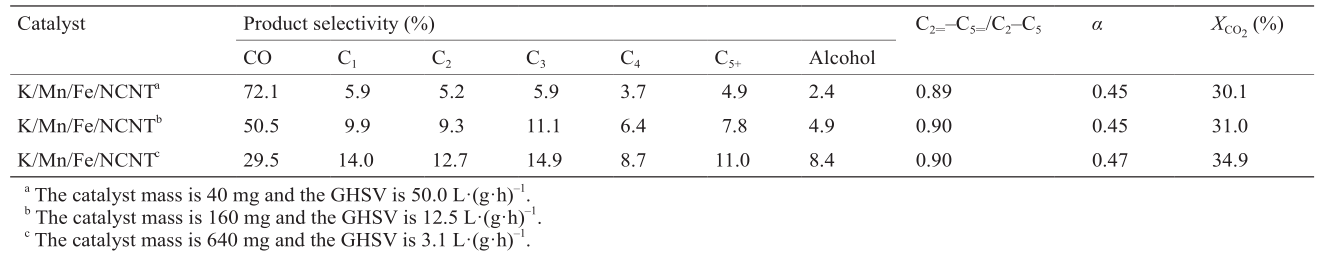
通过K助剂处理的催化剂可实现CO2加氢过程高的烯烃和乙醇选择性。FTS过程生成碳氢化合物的机理是在活性Fe表面吸附的CO被还原生成CH2—*,之后进一步生成CH3—CH2—*。如果催化剂的表面为极性,存在K+,其可以吸取CH3—CH2—*中的氢离子,从而生成CH2=CH2。因此K助剂处理的催化剂将带来较高的烃类选择性,但是K/Fe/NCNT催化剂较容易结焦,在较长的运行时间内表现出活性不稳定。K助剂处理的催化剂可能会吸附水,使得部分亚甲基氧化形成羟基,双助剂处理得到的K/Mn/Fe/NCNT作为除此之外的结构化助剂显示出更加稳定的性能。我们之后的工作将集中于优化K/Mn助剂的用量。这些研究将包括S作为新加入的助剂,其可以非常有效地结合Na元素。
《4.结论》
4.结论
本文研究了在25 bar压力下CO2加氢过程中K助剂和Mn助剂对于Fe/NCNT催化剂的影响。焙烧阶段催化剂主要为Fe3O4,而在CO2加氢过程中其全部转化为了χ-Fe5C2。双助剂处理过后的催化剂K/Mn/Fe/NCNT在反应过后,除了主要铁相χ-Fe5C2之外,还存在一些MnCO3。TPR和原位XANES显示,K助剂降低了催化剂的还原性。Mn助剂稳定了FeO,降低了TPR分析的起始温度。NH3分解被用于探针反应,发现双助剂处理的催化剂K/Mn/Fe/NCNT发挥了最高的催化活性和热稳定性。同时K/Mn/Fe/NCNT显示出很好的费托过程活性,表现为具有较高的CO2转化率、较低的烃类选择性、较高的短链烯烃选择性。在CO2转化率为34.9%的同时具有较高的烯烃/烷烃比率,同时在体积空速为3.1L•(g•h)–1时,CO的选择性可达29.5%。实验结果清楚地显示了K助剂作为电子助剂在铁基纳米颗粒表面化学改性中的作用,而Mn助剂作为结构助剂抑制了烧结现象,提高了催化剂的稳定性。
《Acknowledgements》
Acknowledgements
This work is supported by the Synchrotron Light Research Institute (Public Organization), Thailand (GS- 54-D01) and the Commission on Higher Education, Ministry of Education, Thailand, and was performed under the project“Sustainable Chemical Synthesis (SusChemSys),” which is co-financed by the European Regional Development Fund (ERDF) and the state of North Rhine-West- phalia, Germany, under the Operational Programme“Regional Competitiveness and Employment”2007–2013.
《Compliance with ethics guidelines》
Compliance with ethics guidelines
Praewpilin Kangvansura, Ly May Chew, Chanapa Kongmak, Phatchada Santawaja, Holger Ruland, Wei Xia, Hans Schulz, Attera Worayingyong, and Martin Muhler declare that they have no conflict of interest or financial conflicts to disclose.











 京公网安备 11010502051620号
京公网安备 11010502051620号




