

《1、 引言》
1、 引言
均相反应在化学动力学角度是高效反应,在化学合成领域被广泛采用[1‒3]。例如,一般认为均相催化的反应速率要高于非均相催化。因此确定合适的溶剂以溶解反应物是进行反应的第一步[4‒6],这是因为反应物分子在热溶液或均相中容易表现出较高的活性,在均相状态下,充分分散的反应物分子之间可频繁发生有效碰撞[7‒9]。反应同时也受到热力学规律的制约,只有当这些反应在热力学角度被证明是可行的,才有可能实现。
上述原理体现在共聚物的合成中,特别是对于在室温条件下以固相形式存在的反应物或单体之间的共聚反应。此外,有无数种潜在化学品可作为共聚的反应物或单体,其中许多反应物或单体具有较大的分子量,它们只有在适当的溶剂中溶解时才表现出较高的活性[10‒11]。此外,赋予同系共聚物以不同性质或多种功能是目前研究的趋势[12‒14]。由于反应类型相同,只要可聚合的官能团保持不变,就可以自由选择、设计或修饰同系物单体[15‒16]。因此,同类型聚合所需的溶剂和温度等反应条件往往只需要很小的改变。例如,Hu等[17]总结了21种可供聚合的甲基丙烯酸酯衍生物,这些衍生物可用于制备在水/甲醇或四氢呋喃(THF)中具有不同pH值响应性的叔胺甲基丙烯酸酯基嵌段共聚物。然而,这份可供使用的同系衍生物名单并非无限长,因为基于某些特定的同系衍生物所预期的反应在化学动力学和热力学角度可能是不可行的。
本研究就涉及了一种理想但“不可行”的反应:1,3,5-三(氯甲基)-2,4,6-三甲基苯(TTB)与三聚氰胺的共聚。此反应的预期共聚物可在特定条件下碳化,有望作为制备氮掺杂多孔碳(NDPC)的理想前驱体。NDPC在催化、储能、电化学和吸附等多个领域均已显示出巨大的应用潜力[18‒21]。可以证明聚合物前驱体如果具有相对较高的芳香性和氮含量,就有利于后续NDPC的碳产率、孔隙发育和残氮含量[22‒23],因此通常认为芳香烃和含氮有机物是制备此类聚合物前驱体的理想单体。比如将芳酰卤化物与对苯二胺、乙二胺或3,5-二氨基-1,2,4-三唑聚合。这些霍夫曼型共聚可以在特定的溶剂中顺利实现,反应效率高,以至根本无需使用催化剂[24‒25]。
在本研究开始时,发现在6种溶剂中TTB都完全不能与三聚氰胺反应。通过第一性原理计算,证实了无论在真空条件下还是在溶剂条件下,预期的共聚反应在热力学和动力学上都是不可行的,然而采用无溶剂法却成功实现了TTB和三聚氰胺的共聚。这一异常现象通过实验确认,并通过第一性原理计算和分子动力学(MD)模拟进行解释,结果表明无溶剂法和溶剂法的共聚反应的过程完全不同。实施这种“不可行”反应是成功的,由该反应得到的共聚物展现出构筑具有大比表面积和良好孔道结构的NDPC的理想潜力。后续得到的NDPC具有理想的CO2捕获能力,且对CO2具有良好的吸附选择性和可重复使用性,尤其是700 ℃下碳化的NDPC在0 ℃和1 bar(1 bar = 105 Pa)时CO2吸附量为6.4 mmol·g-1,这一数值可与许多最近报道的NDPC竞争,如WSC-500-1、NPC-1-700和KBM-900 (0 ℃和1 bar时CO2吸附量分别为6.0 mmol·g-1、5.1 mmol·g-1和4.4 mmol·g-1)。
《2、 材料与方法》
2、 材料与方法
《2.1 材料合成》
2.1 材料合成
共聚反应设计方案如图1所示。所有化学品均商业购买并原样使用。在N2气氛下,TTB(5.0 mmol)和三聚氰胺(5.0 mmol)溶于特定溶剂(50 mL)中,加热回流72 h。本研究选择甲醇、THF、环己烷和N,N-二甲基甲酰胺(DMF)作为溶剂,反应温度分别设为65 ℃、70 ℃、80 ℃和155 ℃;然后选择二甲亚砜(DMSO)和乙二醇作为溶剂,在0.5 MPa的N2气氛下,210 ℃在高压釜中反应72 h(210 ℃也是下述无溶剂法的反应温度)。
《图1》
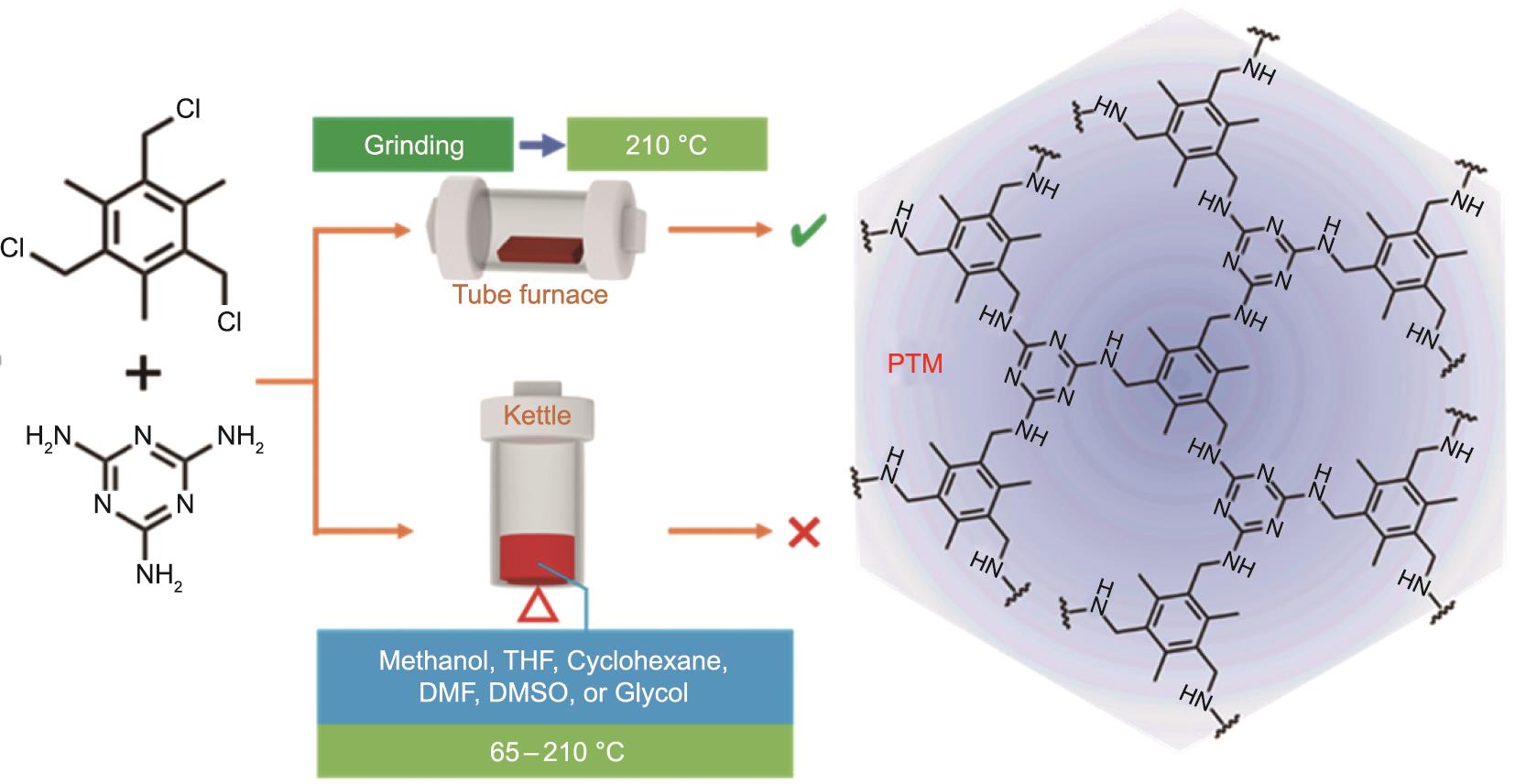
图1 溶剂法与无溶剂法合成PTM方案。
无溶剂法中,采用NaCl粉末(0.06 g)作为助磨剂,将TTB(5.0 mmol)和三聚氰胺(5.0 mmol)在玛瑙研钵中研磨15 min [26‒28]。N2保护下,将混合物转入管式炉,210 ℃加热维持24 h,得到棕色共聚物粉末,然后用甲醇回流纯化3 h以去除未反应的单体,然后用甲醇、CH2Cl2、去离子水依次洗脱,进一步去除残留的单体和NaCl。在100 ℃下真空干燥24 h,最终得到TTB和三聚氰胺的共聚物,代号PTM。
PTM衍生的NDPC(代号PTMC-x,其中x表示碳化温度)的制备过程如下。PTM分散在含有KOH(质量分数为30%)的乙醇/水(1/1,V/V)溶液中。充分浸渍并蒸发溶剂后,固态混合物分别在规定温度(500 ℃、600 ℃、700 ℃、800 ℃)进行N2保护,碳化60 min。产物用2 mol·L-1 HCl溶液洗涤以去除残留的KOH,后用去离子水洗涤,在120 ℃真空中干燥24 h,最后得到PTMC-x。
《2.2 实验表征》
2.2 实验表征
采用Nicolet Nexus 470光谱仪(KBr晶片)记录傅里叶变换红外光谱(FTIR;Nicolet iS10,美国)。采用Agilent-NMR-vnmrs600光谱仪(美国)记录固态13C核磁共振(13C NMR)谱。用Bruker D8 Advance衍射仪记录40 kv和40 mA下Cu Kα辐射的X射线粉末衍射(XRPD;Bruker D8 Advance,德国)图。元素分析采用德国Elementar Vario EL元素分析仪进行。扫描电子显微镜(SEM)图像使用日立S-4800和日本FEI Tecnai G2T20电子显微镜获得。材料的透射电子显微镜(TEM)图像用JEM-2010 UHR电子显微镜(日本)在200 kV下拍摄。用HR800紫外拉曼微光谱仪(HORIBA,美国)记录拉曼光谱。X射线光电子能谱(XPS)使用ESCALAB-220I-XL装置(VG Scientific,英国)进行。使用Micromeritics ASAP 2020分析仪(Micromeritics,美国)测量150 ℃脱气4 h后的样品于77 K下的N2吸脱附等温线,并利用吸附等温线的数据计算Brunauer-Emmett-Teller(BET)比表面积,由相对压力为0.95时的吸附量推断总孔体积。孔径分布则采用非局域密度泛函理论(NLDFT)估计。在0 ℃和25 ℃条件下,使用ASAP 2020分析仪分别对吸附剂进行CO2(99.999%)和N2(99.999%)的静态吸附实验。自由空间由氦气(99.999%)确定,并假设氦气在测试温度下未被吸附。
《3、 结果与讨论》
3、 结果与讨论
《3.1 共聚反应机理》
3.1 共聚反应机理
虽然已经证明霍夫曼型共聚是足够高效的,但本研究通过实验证实,使用6种代表性溶剂中的任何一种,即甲醇、THF、环己烷、DMF、DMSO和乙二醇,都不会使TTB和三聚氰胺生成共聚产物。正如使用第一原理计算所得(计算细节见附录A)[29‒32],TTB和三聚氰胺之间的反应在热力学或动力学角度是被禁阻的。如图2(a)所示,TTB和三聚氰胺之间的反应一步即可完成,表现出典型的SN2反应特征。TTB的芳香环与三聚氰胺的氮杂环平行,这是二者的有效碰撞方向,因此三聚氰胺的一个氨基抵迫TTB的—Cl基团,使Cl原子被剥离,同时生成了过渡态(TS)。活化的Cl原子立即抓取同样被抵迫的氨基H原子,在形成—CH2—NH—键的同时生成HCl分子。该反应在298 K时在真空中的内禀自由能变化为4.5 kcal·mol-1(1 kcal=4.18 kJ),根据热力学定律可知,在环境温度下此反应是热力学禁阻的。同时,计算得到的真空反应能垒达到37.2 kcal·mol-1,除非反应温度能适当升高,否则此能垒也足以抑制反应进行。
《图2》

图2 (a)第一原理计算的在真空和甲醇反应场中共聚反应的势能面;(b)随温度变化的共聚反应自由能变化;(c)反应速率常数的对数与温度倒数之间的关系;(d)MD模拟的0 K和483 K下反应物分子在50 ps时的相对位移;(e)MD模拟的0 K和338 K下100个甲醇分子包围的一对反应物分子在50 ps时的相对位移。
本研究进而基于极化连续介质模型模拟反应物附近的甲醇溶剂分子存在的影响。计算结果表明,即使存在甲醇溶剂的影响,TTB和三聚氰胺之间的相对位置也不会发生显著变化。如图2(a)所示,反应的自由能变化(2.9 kcal·mol-1)和反应能垒(27.5 kcal·mol-1)都由于甲醇的影响而有所降低,然而在298 K下这仍然是一个禁阻反应。此外,使用吉布斯-亥姆霍兹方程(Gibbs-Helmholtz equation),计算了在298 K至498 K的温度范围内,反应在真空和甲醇中的自由能变化,这个温度范围涵盖了本研究中尝试共聚的各个操作温度[图2(b)]。研究表明,在所有目标温度下,反应的自由能变化均为正值,说明在本研究所有的溶剂中和相对较高温度下此反应的不可行性。图2(c)显示了温度对反应速率常数k的影响。随着温度升高,k值迅速增加。例如,甲醇中反应的k值从298 K的3.89 × 10-8 s-1升为338 K的7.60 × 10-6 s-1。当温度升高到483 K时,反应的k值达到2.25 × 10-5 s-1,这是210 °C下无溶剂法的理论k值。k值的增加表明反应在化学动力学方面更容易发生,而正的自由能变化值却表明反应不能自发发生,除非无溶剂法打破这种平衡。值得注意的是,HCl是共聚的副产物,而DMSO或乙二醇作为溶剂时的操作温度与无溶剂法相同。与溶剂法不同,无溶剂法立即将生成的HCl释放到大气中,从而使产物的复合结构解离。因此,TTB和三聚氰胺之间的反应被推向正反应方向。
MD模拟表明了该反应仅在使用无溶剂法时发生的另一个重要原因——固相。TTB和三聚氰胺(1∶1)混合物的单胞参数的MD模拟结果与第一原理计算结果一致,表明MD模拟的准确性和可靠性(附录A,表S1)。如图2(d)所示,在483 K下对27个TTB和27个三聚氰胺分子进行50 ps的MD模拟,以描述系统在实验温度下的热运动,该温度对于激活反应来说已足够高,但仅略微改变了单体分子在固相中的相对位置。因此,TTB和三聚氰胺分子可以相互平行碰撞并引发反应。相较而言,分散在甲醇分子中的单体分子却受到显著干扰。如图2(e)所示,当一对TTB和三聚氰胺分子与100个甲醇分子共存时,338 K下50 ps的MD模拟显示TTB和三聚氰胺分子会被甲醇分子分隔开。此外,—NH2和—Cl基团之间的距离从0 K时的3.5 Å延伸到338 K时的7.9 Å,这个距离太长,无法引发反应。单体分子之间有效距离的重要性被另一个显著的实验事实证明——如果反应温度升高到220 ℃以上,混合物中的TTB开始熔化,单体分子的活性自会显著增加,但此时即使利用无溶剂法也不会生成共聚物产物。这种情形与溶剂环境类似,熔融相中过度活化的TTB分子会与三聚氰胺分子过度分离。
上述理论和实验结果揭示了使用无溶剂法成功共聚TTB和三聚氰胺的两个根本原因——及时释放的副产物HCl所推动的反应平衡,以及被限制在固相中的活化单体分子的热运动。此外,三聚氰胺的热缩合在无溶剂反应过程中也发挥了一定作用,可以确保最终NDPC产物的稳定性。
《3.2 共聚物表征》
3.2 共聚物表征
PTM的成功制备由图3(a)中FTIR谱证实。位于3348 cm-1和3216 cm-1处的谱带归因于—NH—,位于2916 cm-1处的谱带是由烷基C—H的拉伸振动引起的。1682 cm-1和1643 cm-1处的强谱带表示存在芳香环,该谱带被由1547 cm-1处—NH—的面内弯曲振动引起的最强谱带所掩蔽。在1458 cm-1和1335 cm-1处的谱峰分别归因于共聚过程中产生的—CH2—基团和连接至芳香环的—CH3基团的面内弯曲振动。在指纹区,1169 cm-1和1022 cm-1处的两个谱带由共聚过程中形成的脂肪族C—N键的拉伸振动引起。两个弱峰位于810 cm-1和780 cm-1处,二者归因于三聚氰胺片段中存在C—N键,并且—NH—的振动在690 cm-1处出峰。13C NMR谱为共聚物的形成提供了更多证据。如图3(b)所示,位于38.6 ppm和72.3 ppm处的谱峰表明新形成了—CH2—NH—键,13.2 ppm处的峰对应于—CH3基团的C原子。130.8 ppm和162.9 ppm处的两个峰由两种类型的芳香族C原子引起,而187.7 ppm处的峰归因于三嗪环的C原子。根据FTIR和13C NMR的表征结果,TTB和三聚氰胺之间的反应主要是单取代反应,即形成—CH2—NH—键,这是因为无溶剂法未使用催化剂,并且本研究基于目标聚合物的理论结构采用了严格的反应物摩尔比。在之前的报道[23‒24,33]中,该方法已被证明是可靠的。
《图3》

图3 PTM 的FTIR谱(a)和13C NMR谱(b)。
元素分析结果进一步佐证了PTM的成功制备。如表1所示,C、H和N的元素含量分别为63.9%、6.6%和27.5%,这与理论值(C, 63.8%; H, 6.4%; N, 29.8%)基本一致。通过差异归一化,其他元素含量主要归因于TTB单体中的O和痕量Cl的存在。由于可旋转或柔性σ型—CH2—NH—键的存在,本研究合成的PTM被确认为非晶态,这是因为PTM的大分子可以在聚合物链增长过程中随机折叠和卷曲。图S1和图S2(附录A)中XRPD和SEM图像显示了PTM具有典型的非晶态特征。基于7个TTB和15个三聚氰胺分子共聚形成的PTM分子片段,第一原理计算模拟了PTM在分子水平上的形态(附录A,图S3)。由于可旋转的σ型—CH2—NH—键和较强的分子内范德华力,PTM分子片段折叠和卷曲到分子收缩的程度。作为NDPC的理想前驱体,PTM折叠和卷曲的构型导致芳香环和三嗪环之间的距离较短。一旦形成自由基或其他活性中间体,PTM很容易自缩合,同时N原子很容易掺杂到碳微晶中,形成具有理想组织特性和应用性能的NDPC。
《表1》
表1 PTM和PTMC的结构参数和元素分析.
| Sample | Yield (wt%) | Elemental content (wt%) | ||||||
|---|---|---|---|---|---|---|---|---|
| C | H | N | Others | |||||
| PTM | 82.1 | 18 | 0.05 | — | 63.9 | 6.6 | 27.5 | 2.0 |
| PTMC-500 | 36.2 | 1102 | 0.58 | 0.51 | 78.9 | 6.5 | 8.3 | 6.3 |
| PTMC-600 | 32.4 | 1809 | 0.73 | 0.70 | 81.4 | 4.7 | 7.1 | 6.8 |
| PTMC-700 | 27.3 | 2603 | 1.16 | 1.03 | 85.3 | 2.5 | 4.8 | 7.4 |
| PTMC-800 | 15.2 | 1442 | 0.66 | 0.57 | 91.1 | 1.7 | 2.5 | 4.7 |
《3.3 PTM生成的NDPC》
3.3 PTM生成的NDPC
PTM通过碳化可衍生出一系列NDPC,代号为PTMC。如图S4(附录A)所示,随着碳化温度升高,FTIR谱上PTM的特征谱带逐渐消失,表明随着官能团的持续解离,碳微晶不断生成。此外,拉曼光谱证明了碳质结构的演变。如图S5(附录A)所示,两个以1345 cm-1和1581 cm-1为中心的特征峰分别对应于无序结构或碳质微晶缺陷的D带和表示sp2碳原子的面内振动的G带。随着碳化温度从500 ℃升高到800 ℃,D/G带的强度比不断降低,表明NDPC的石墨化程度增加。PTMC的XRPD图(附录A,图S6)无明显尖锐衍射峰,呈现典型的非晶态特征。位于25°和43°处的两个弥散衍射峰分别归因于NDPC的(002)和(100)石墨晶面的反射。随着碳化温度升高,两个弥散衍射峰从背景中凸显,表明NDPC的石墨化程度进一步发展,这与拉曼光谱结果一致。从表1可以看出,PTM展现出产生NDPC的良好潜质,具有令人满意的碳产率。例如,据了解,PTMC-700的27.3%(质量分数)的碳产率是用其他人工聚合物前驱体很难达到的。根据XPS结果(附录A,图S7),在NDPC上存在三种N掺杂位点——吡咯氮(N-5)、吡啶氮(N-6)和四元氮(N-Q)[24]。当碳化温度从500 ℃提高到800 ℃时,NDPC的残氮含量与其石墨化程度相互折衷,而N-5位点比其他两种氮掺杂位点更稳定。因此,NDPC上保留了更多的N-5位点。PTMC的总氮含量,例如,PTMC-700为4.8%与其他报道中的一些代表性NDPC的氮含量相比也颇具优势(附录A,表S2)。
PTM作为NDPC前驱体最显著的优势体现在所生成的PTMC的结构特性上,其结构特性可使用N2吸脱附等温线进行研究,计算得到的结构参数如表1所示。如图S8(a)(附录A)所示,所有PTMC均具有典型的I型等温线,在低P/P0范围内可快速吸附N2,表明PTMC存在大量微孔。所有PTMC的吸脱附等温线回滞环都不明显,表明其少量介孔结构即使存在,也是由颗粒堆积引起的。PTM具有很少的孔,其BET比表面积相当低,原因如前所述,作为NDPC的前驱体,PTM是无定形和致密的。然而,PTMC的BET比表面积在碳化后足够高。与其他PTMC(PTMC-500: 1102 m2·g-1; PTMC-600: 1809 m2·g-1; PTMC-800: 1442 m2·g-1)相比,PTMC-700的具有最高BET比表面积(2603 m2·g-1),这个值也高于许多其他报道的NDPC(见附录A中的表S2)。PTMC-800的BET比表面积降低是由于过度碳化导致碳微晶进一步分解。BET比表面积的变化趋势与PTMC的孔隙率的变化趋势一致,如附录A中图S8(b)和表1所示,PTMC的孔体积随碳化温度升高而增加,直到碳化温度达到700 ℃(1.16 cm3·g-1),而其微孔体积也同步增加至1.03 cm3·g-1。对于每个PTMC,超过85%的孔体积均由微孔贡献。此外,在SEM图像中可观察到PTMC孔结构的变化(附录A,图S9)。可以证明,500 ℃的温度已足以碳化PTM(如FTIR光谱所示),而PTMC-500的孔结构尚不发达。如PTMC-700的颗粒横截面所示,随着碳化温度升高到600 ℃、700 ℃,PTMC的孔结构逐步发达。800 ℃的过度碳化导致PTMC-800的碳组织结构瓦解,此时其BET比表面积和孔体积均减小。
较大的BET比表面积和良好的孔结构通常表明该材料在许多应用领域(如气体储存和分离)具有良好的潜力。因此,在本研究中,PTMC被用作吸附剂来捕获CO2。如图4所示,与其他PTMC相比,PTMC-700表现出更高的CO2捕获能力,其CO2吸附容量为6.4 mmol·g-1(0 ℃、1 bar)和3.4 mmol·g-1(25 ℃、1 bar),因为PTMC-700具有最大的BET比表面积和孔体积,特别是微孔体积,而微孔被证明是NDPC得以限域CO2分子的关键。此外,如附录A中的表S2所示,PTMC-700不仅表现出更高的BET比表面积,而且与其他报道的代表性NDPC [34‒45],如WSC-500-1、NPC-1-700和KBM-900(在0 ℃和1 bar下分别为6.0 mmol·g-1、5.1 mmol·g-1和4.4 mmol·g-1)相比,其CO2捕获能力也具有竞争力。理想吸附溶液理论(附录A,图S10和图S11)证实了PTMC对CO2/N2的良好选择性。PTMC-700的CO2/N2(85/15, V/V)选择性值在0 ℃和25 ℃时分别达到18和32,与其他一些代表性NDPC的选择性值相当(见附录A中的表S2)。这在很大程度上取决于PTMC-700存在足够的氮掺杂位作为CO2分子的亲和位点。值得注意的是,由于碳化,一般NDPC的残氮含量普遍较低,而在本研究中,PTMC-700的残氮含量达到4.8%,高于其他代表性NDPC(见附录A中的表S2)。
《图4》

图4 PTMC在0 ℃和25 ℃下的CO2吸附等温线。
NDPC的CO2吸附性能主要取决于三个因素的协同效应——微孔、氮掺杂位点和比表面积[23]。微孔提供了限域CO2分子的微环境,NDPC中掺杂的氮物种有利于CO2锚定,原因是氮物种可以在吸附剂表面和CO2分子之间产生较强的范德华力[46],后续可增强CO2吸附量和选择性。在本研究中,由于与BET比表面积的一致性关系,NDPC的孔结构主导了对CO2的捕获,然而仍然可以观察到氮掺杂位点的显著贡献,使得PTMC-500的CO2/N2选择性高于PTMC-800(附录A,图S11)。
此外,利用计算得到的等量吸附热值(Qst)可证明CO2分子和NDPC之间为物理吸附,基于0 ℃和25 ℃下的CO2吸附等温线数据得到的所有Qst值均低于30 kJ·mol-1(附录A,图S12)。可重复使用性是评价吸附剂的另一个重要指标。为了检测可重复使用性,PTMC-700在130 ℃下抽真空100 min进行再生,然后在0 ℃下用CO2进行饱和吸附至1 bar。如附录A中图S13所示,即使在6次CO2吸脱附循环后,也未观察到PTMC-700的CO2捕获能力衰减。在重复使用性测试后,对PTMC-700进行了N2吸脱附测试和孔分布分析,以研究其组织特性的变化(附录A,图S14)。实验发现PTMC-700的BET比表面积仍达到2600 m2·g-1,总孔体积为1.16 cm3·g-1,并且孔径分布保持良好。这进一步证实了PTMC-700对于CO2吸附具有良好的可重复使用性。令人满意的NDPC碳产量、具有竞争力的残氮含量和结构特性、良好的CO2捕获能力和选择性,以及完美的可重复使用性共同证明了PTM产生的PTMC符合预期。此外也证明,通过无溶剂法制备的PTM的确是NDPC的良好前驱体。
作为本研究的优势之一,该方法论提供了一种潜在的渠道以实现那些在溶剂环境中“不可行”的霍夫曼烷基化反应。此外,根据之前的报道[47],基于霍夫曼烷基化反应的一些目标材料的合成必须在溶剂中使用腐蚀性强的碱性催化剂,并且产物的纯化过程很复杂。因此,本研究的另一个优势是,采用无溶剂法,无需腐蚀性催化剂,并且产物的纯化比溶剂法容易得多。此外,利用本研究中提出的方法,可以实施某些共聚反应,以制备具有特定分子结构的前驱体,以促进后续多孔碳孔结构的生成,或者制备含有特定待嫁接基团的聚合物,后续可被特定的含氮基团(如氨基)取代[34]。理论上,如此即可独立调节后续多孔碳产品的孔结构和氮掺杂浓度。
《4、 结论》
4、 结论
本研究利用无溶剂法成功实现了1,3,5-三(氯甲基)-2,4,6-三甲基苯和三聚氰胺的共聚,该共聚反应不能在溶剂中进行。第一性原理计算证明,该反应在化学动力学和热力学方面是不可行的。然而,无溶剂合成过程中副产物HCl的迅速释放和足够高的反应温度分别突破了热力学和动力学的限制,这在溶剂环境中是无法实现的。此外MD模拟表明,一个紧致的反应物分子复合结构对于实施该反应至关重要,此类结构可以在无溶剂法的固相中实现;而在溶剂环境中,溶剂分子分隔反应物分子,从而抑制了反应物分子之间的有效碰撞。制备这种具有高芳香性和高氮含量的共聚物的努力被证明是值得的。通过共聚物的碳化,生成了一系列具有令人满意的碳产率、高残氮含量、良好的孔结构和足够高的BET比表面积的NDPC。由于孔结构和氮掺杂位点的完美协同,这些NDPC表现出很高的CO2捕获能力、良好的CO2选择性和理想的可重复使用性,尤其是在700 ℃下碳化得到的NDPC。这些特性使本研究中的NDPC与其他报道中的同类吸附剂相比更具有竞争力。本研究中展示的共聚反应的潜在机制也启发人们对其他一些“不可行”但期望发生的反应进行重新审视。











 京公网安备 11010502051620号
京公网安备 11010502051620号




