
2017, Volume 3, Issue 3
Engineering >> 2017, Volume 3, Issue 3 doi: 10.1016/J.ENG.2017.03.013
Effects of Potassium and Manganese Promoters on Nitrogen-Doped Carbon Nanotube-Supported Iron Catalysts for CO2 Hydrogenation
a Scientific Equipment Center, Faculty of Science, Kasetsart University, Bangkok 10900, Thailand
b Laboratory of Industrial Chemistry, Ruhr-University Bochum, Bochum 44780, Germany
c Department of Materials Science, Faculty of Science, Kasetsart University, Bangkok 10900, Thailand
d Department of Chemistry, Faculty of Science, Kasetsart University, Bangkok 10900, Thailand
e Engler-Bunte-Institute, Karlsruhe Institute of Technology, Karlsruhe 76131, Germany
Next Previous
Abstract
Nitrogen-doped carbon nanotubes (NCNTs) were used as a support for iron (Fe) nanoparticles applied in carbon dioxide (CO2) hydrogenation at 633 K and 25 bar (1 bar = 105 Pa). The Fe/NCNT catalyst promoted with both potassium (K) and manganese (Mn) showed high performance in CO2 hydrogenation, reaching 34.9% conversion with a gas hourly space velocity (GHSV) of 3.1 L·(g·h)−1. Product selectivities were high for olefin products and low for short-chain alkanes for the K-promoted catalysts. When Fe/NCNT catalyst was promoted with both K and Mn, the catalytic activity was stable for 60 h of reaction time. The structural effect of the Mn promoter was demonstrated by X-ray diffraction (XRD), temperature-programmed reduction (TPR) with molecular hydrogen (H2), and in situ X-ray absorption near-edge structure (XANES) analysis. The Mn promoter stabilized wüstite (FeO) as an intermediate and lowered the TPR onset temperature. Catalytic ammonia (NH3) decomposition was used as an additional probe reaction for characterizing the promoter effects. The Fe/NCNT catalyst promoted with both K and Mn had the highest catalytic activity, and the Mn-promoted Fe/NCNT catalysts had the highest thermal stability under reducing conditions.
Keywords
CO2 hydrogenation ; Iron catalyst ; Nitrogen-doped carbon nanotubes ; Manganese promoter ; Potassium promoter
Figures
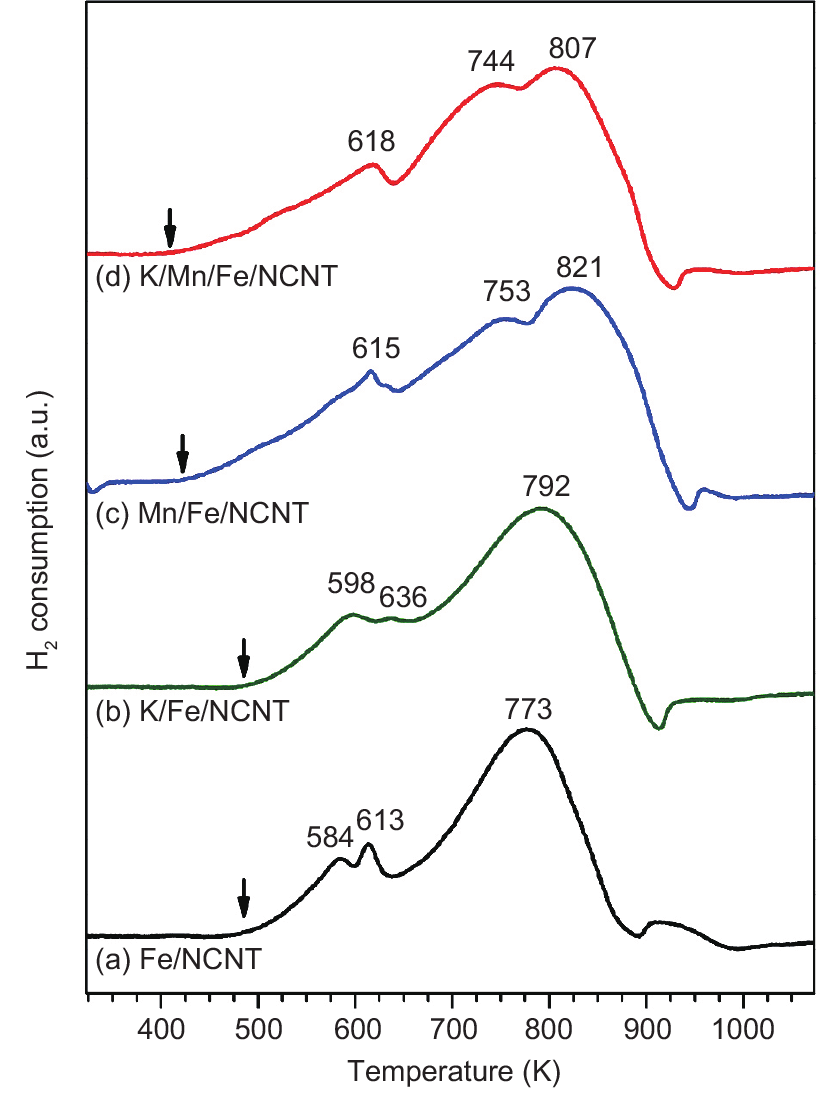
Fig. 1

Fig. 2
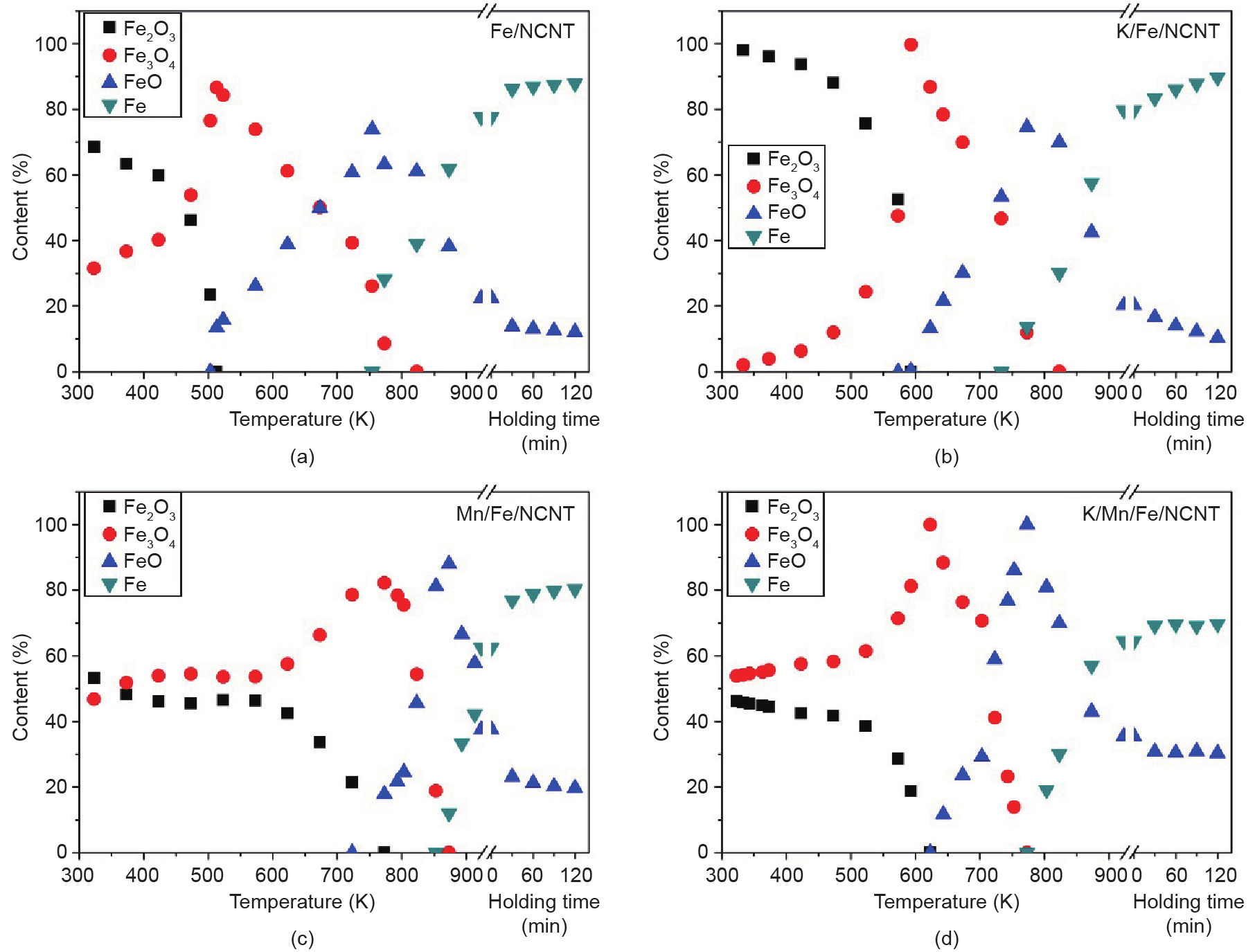
Fig. 3
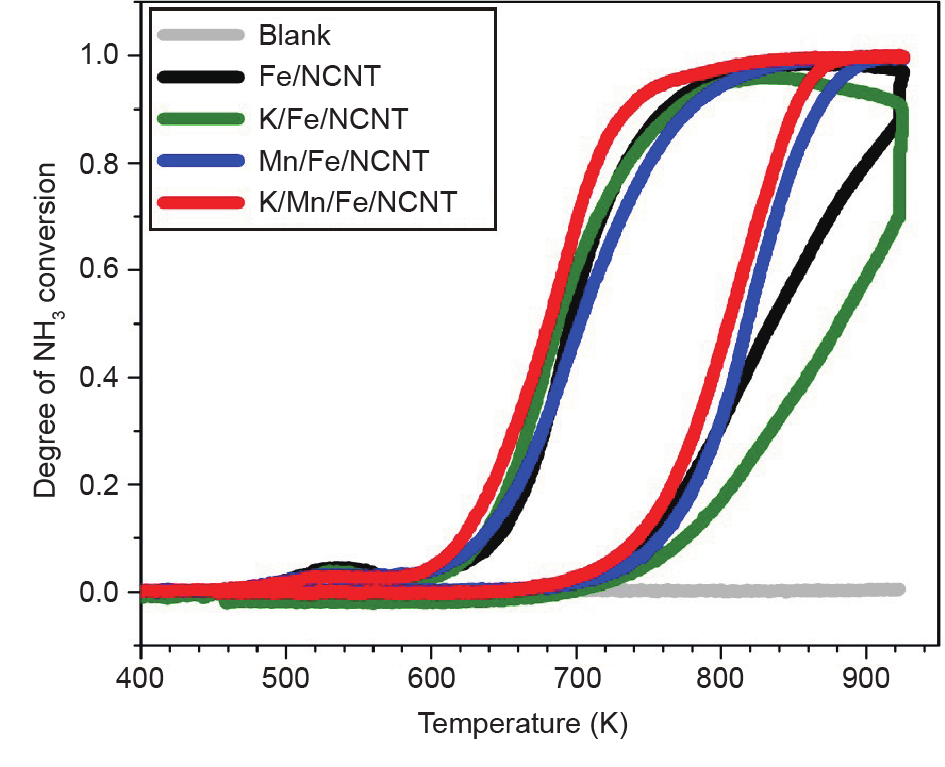
Fig. 4

Fig. 5
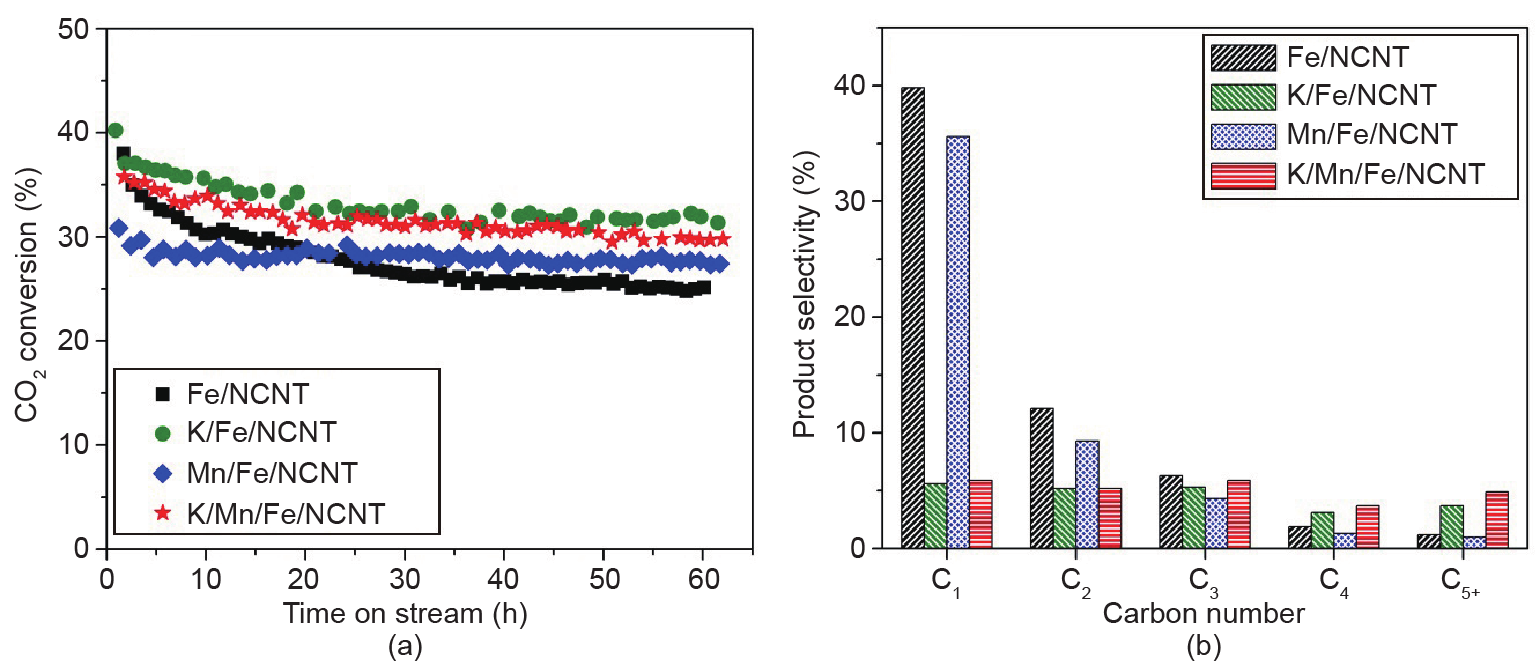
Fig. 6

Fig. 7
References
[ 1 ] Wang W, Wang S, Ma X, Gong J. Recent advances in catalytic hydrogenation of carbon dioxide. Chem Soc Rev 2011;40(7):3703–27 link1
[ 2 ] Chew LM, Kangvansura P, Ruland H, Schulte HJ, Somsen C, Xia W, et al.Effect of nitrogen doping on the reducibility, activity and selectivity of carbon nanotube-supported iron catalysts applied in CO2 hydrogenation. Appl Catal A Gen 2014;482:163–70 link1
[ 3 ] Schulz H, Riedel T, Schaub G. Fischer-Tropsch principles of co-hydrogenation on iron catalysts. Top Catal 2005;32:117–24 link1
[ 4 ] Schulz H. Comparing Fischer-Tropsch synthesis on iron- and cobalt catalysts: The dynamics of structure and function. Stud Surf Sci Catal 2007;163:177–99 link1
[ 5 ] Abbaslou RMM, Tavassoli A, Soltan J, Dalai AK. Iron catalysts supported on carbon nanotubes for Fischer-Tropsch synthesis: Effect of catalytic site position. Appl Catal A Gen 2009;367(1–2):47–52 link1
[ 6 ] Riedel T, Schulz H, Schaub G, Jun KW, Hwang JS, Lee KW. Fischer-Tropsch on iron with H2/CO and H2/CO2 as synthesis gases: The episodes of formation of the Fischer-Tropsch regime and construction of the catalyst. Top Catal 2003;26(1):41–54 link1
[ 7 ] Riedel T, Claeys M, Schulz H, Schaub G, Nam SS, Jun KW, et al.Comparative study of Fischer-Tropsch synthesis with H2/CO and H2/CO2 syngas using Fe- and Co-based catalysts. Appl Catal A Gen 1999;186(1–2):201–13 link1
[ 8 ] Srinivas S, Malik RK, Mahajani SM. Fischer-Tropsch synthesis using bio-syngas and CO2. Energy Sustain Dev 2007;11(4):66–71 link1
[ 9 ] Chen W, Fan Z, Pan X, Bao X. Effect of confinement in carbon nanotubes on the activity of Fischer-Tropsch iron catalyst. J Am Chem Soc 2008;130(29):9414–9 link1
[10] de Smit E, Beale AM, Nikitenko S, Weckhuysen BM. Local and long range order in promoted iron-based Fischer-Tropsch catalysts: A combined in situ X-ray absorption spectroscopy/wide angle X-ray scattering study. J Catal 2009;262:244–56 link1
[11] Pour AN, Housaindokht MR, Tayyari SF, Zarkesh J. Fischer-Tropsch synthesis by nano-structured iron catalyst. J Nat Gas Chem 2010;19(3):284–92 link1
[12] Pour AN, Housaindokht MR, Tayyari SF, Zarkesh J. Deactivation studies of nano-structured iron catalyst in Fischer-Tropsch synthesis. J Nat Gas Chem 2010;19(3):333–40 link1
[13] de Smit E, Cinquini F, Beale AM, Safonova OV, van Beek W, Sautet P, et al.Stability and reactivity of ε-χ-θ iron carbide catalyst phases in Fischer-Tropsch synthesis: Controlling μ c. J Am Chem Soc 2010;132(42):14928–41 link1
[14] Xiong H, Moyo M, Motchelaho MA, Jewell LL, Coville NJ. Fischer-Tropsch synthesis over model iron catalysts supported on carbon spheres: The effect of iron precursor, support pretreatment, catalyst preparation method and promoters. Appl Catal A Gen 2010;388(1–2):168–78 link1
[15] Yu G, Sun B, Pei Y, Xie S, Yan S, Qiao M, et al.FexOy@C spheres as an excellent catalyst for Fischer-Tropsch synthesis. J Am Chem Soc 2010;132(3):935–7 link1
[16] Dorner RW, Hardy DR, Williams FW, Willauer HD. Heterogeneous catalytic CO2 conversion to value-added hydrocarbons. Energy Environ Sci 2010;3(7):884–90 link1
[17] Dorner RW, Hardy DR, Williams FW, Willauer HD. K and Mn doped iron-based CO2 hydrogenation catalysts: Detection of KAlH4 as part of the catalyst’s active phase. Appl Catal A Gen 2010;373(1–2):112–21 link1
[18] Bahome MC, Jewell LL, Hildebrandt D, Glasser D, Coville NJ. Fischer-Tropsch synthesis over iron catalysts supported on carbon nanotubes. Appl Catal A Gen 2005;287(1):60–7 link1
[19] Ribeiro MC, Jacobs G, Davis BH, Cronauer DC, Kropf AJ, Marshall CL. Fischer-Tropsch synthesis: An in situ TPR-EXAFS/XANES investigation of the influence of group I alkali promoters on the local atomic and electronic structure of carburized iron/silica catalysts. J Phys Chem C 2010;114(17):7895–903 link1
[20] Tao Z, Yang Y, Wan H, Li T, An X, Xiang H, et al.Effect of manganese on a potassium-promoted iron-based Fischer-Tropsch synthesis catalyst. Catal Lett 2007;114(3):161–8 link1
[21] Campos A, Lohitharn N, Roy A, Lotero E, Goodwin JG, Spivey JJ. An activity and XANES study of Mn-promoted, Fe-based Fischer-Tropsch catalysts. Appl Catal A Gen 2010;375(1):12–6 link1
[22] Ribeiro MC, Jacobs G, Pendyala R, Davis BH, Cronauer DC, Kropf AJ, et al.Fischer-Tropsch synthesis: Influence of Mn on the carburization rates and activities of Fe-based catalysts by TPR-EXAFS/XANES and catalyst testing. J Phys Chem C 2011;115:4783–92 link1
[23] Davis BH. Fischer-Tropsch synthesis: Reaction mechanisms for iron catalysts. Catal Today 2009;141(1–2):25–33 link1
[24] Torres Galvis HM, Bitter JH, Khare CB, Ruitenbeek M, Dugulan AI, de Jong KP. Supported iron nanoparticles as catalysts for sustainable production of lower olefins. Science 2012;335(6070):835–8 link1
[25] Tavasoli A, Sadagiani K, Khorashe F, Seifkordi A, Rohani A, Nakhaeipour A. Cobalt supported on carbon nanotubes—A promising novel Fischer-Tropsch synthesis catalyst. Fuel Process Technol 2008;89(5):491–8 link1
[26] van Steen E, Prinsloo FF. Comparison of preparation methods for carbon nanotubes supported iron Fischer-Tropsch catalysts. Catal Today 2002;71(3–4):327–34 link1
[27] Schulte HJ, Graf B, Xia W, Muhler M. Nitrogen- and oxygen-functionalized multiwalled carbon nanotubes used as support in iron-catalyzed, high-temperature Fischer-Tropsch synthesis. ChemCatChem 2012;4(3):350–5 link1
[28] Dorner RW, Hardy DR, Williams FW, Willauer HD. Catalytic CO2 hydrogenation to feedstock chemicals for jet fuel synthesis using multi-walled carbon nanotubes as support. In: Hu YH, editor Advances in CO2 conversion and utilization. Washington DC: American Chemical Society; 2010. p. 125–39 link1
[29] Kundu S, Xia W, Busser W, Becker M, Schmidt DA, Havenith M, et al.The formation of nitrogen-containing functional groups on carbon nanotube surfaces: A quantitative XPS and TPD study. Phys Chem Chem Phys 2010;12(17):4351–9 link1
[30] Kowalczyk Z, Sentek J, Jodzis S, Muhler M, Hinrichsen O. Effect of potassium on the kinetics of ammonia synthesis and decomposition over fused iron catalyst at atmospheric pressure. J Catal 1997;169(2):407–14 link1
[31] Arabczyk W, Zamlynny J. Study of the ammonia decomposition over iron catalysts. Catal Lett 1999;60(3):167–71 link1
[32] Kangvansura P, Chew LM, Saengsui W, Santawaja P, Poo-arporn Y, Muhler M, et al.Product distribution of CO2 hydrogenation by K- and Mn-promoted Fe catalysts supported on N-functionalized carbon nanotubes. Catal Today 2016;275:59–65 link1
[33] Xia W, Jin C, Kundu S, Muhler M. A highly efficient gas-phase route for the oxygen functionalization of carbon nanotubes based on nitric acid vapor. Carbon 2009;47(3):919–22 link1
[34] Boot LA, van Dillen AJ, Geus JW, van Buren FR. Iron-based dehydrogenation catalysts supported on zirconia. I. Preparation and characterization. J Catal 1996;163(1):186–94 link1
[35] Poo-arporn Y, Chirawatkul P, Saengsui W, Chotiwan S, Kityakarn S, Klinkhieo S, et al.Time-resolved XAS (Bonn-SUT-SLRI) beamline at SLRI. J Synchrotron Radiat 2012;19(6):937–43 link1
[36] Ravel B, Newville M. ATHENA, ARTEMIS, HEPHAESTUS: Data analysis for X-ray absorption spectroscopy using IFEFFIT. J Synchrotron Radiat 2005;12(4):537–41 link1
[37] Chew LM, Ruland H, Schulte HJ, Xia W, Muhler M. CO2 hydrogenation to hydrocarbons over iron nanoparticles supported on oxygen-functionalized carbon nanotubes. J Chem Sci 2014;126(2):481–6 link1
[38] Wimmers OJ, Arnoldy P, Moulijn JA. Determination of the reduction mechanism by temperature-programmed reduction: Application to small iron oxide (Fe2O3) particles. J Phys Chem C 1986;90(7):1331–7 link1
[39] Pernicone N, Ferrero F, Rossetti I, Forni L, Canton P, Riello P, et al.Wüstite as a new precursor of industrial ammonia synthesis catalysts. Appl Catal A Gen 2003;251(1):121–9 link1
[40] Yeo SC, Han SS, Lee HM. Mechanistic investigation of the catalytic decomposition of ammonia (NH3) on an Fe(100) surface: A DFT study. J Phys Chem C 2014;118(10):5309–16 link1
[41] Jedynak A, Kowalczyk Z, Szmigiel D, Rarog W, Zielinski J. Ammonia decomposition over the carbon-based iron catalyst promoted with potassium. Appl Catal A Gen 2002;237(1–2):223–6 link1
[42] Dad M, Fredriksson H, van de Loosdrecht J, Thuene P, Niemantsverdriet J. Stabilization of iron by manganese promoters in uniform bimetallic FeMn Fischer-Tropsch model catalysts prepared from colloidal nanoparticles. Catal Struct React 2015;1(2):101–9 link1
[43] Grzybek T, Klinik J, Papp H, Baerns M. Characterization of Cu and K containing Fe/Mn oxide catalysts for Fischer-Tropsch synthesis. Chem Eng Technol 1990;14(1):156–61 link1
[44] Lee JF, Chern WS, Lee MD. Hydrogenation of carbon dioxide on iron catalysts doubly promoted with manganese and potassium. Can J Chem Eng 1992;70(3):511–5 link1











 京公网安备 11010502051620号
京公网安备 11010502051620号




