
2019, Volume 5, Issue 4
Engineering >> 2019, Volume 5, Issue 4 doi: 10.1016/j.eng.2019.06.002
Developing a Transformation-Independent and Unbiased qPCR Assay to Rapidly Evaluate the Determinants of DNA Assembly Efficiency
a Key Laboratory of Molecular Medicine and Biotherapy, School of Life Science, Beijing Institute of Technology, Beijing 100081, China
b UCLA Institute for Technology Advancement (Suzhou), Suzhou 215123, China
# These authors contributed equally to this work.
Next Previous
Abstract
Synthetic biology is moving in the direction of larger and more sophisticated design, which depends heavily on the efficient assembly of genetic modules. Conventional evaluation of the DNA assembly efficiency (AE) requires transformation, and the whole process requires up to 10 h and is susceptible to various interferences. To achieve rapid and reliable determination of the AE, an alternative transformation-independent method was established using a modified quantitative polymerase chain reaction (qPCR) assay. The AE is represented by the proportion of the ligated fragment, which can be determined within 3 h. This qPCR-based measurement was tested by the commonly used restriction ligation, Golden Gate assembly, and Gibson assembly for the assembly of two or more DNA pieces; the results correlated significantly with the AEs represented by the counting of the colony-forming units (CFUs). This method outperformed the CFU-based measurement by reducing the measuring bias and the random deviations that stem from the transformation process. The method was then employed to investigate the effects of terminal secondary structures on DNA assembly. The results revealed the major effects of the overall properties of the overlap sequence and the negative effects of hairpin structures on the AE, which are relevant for all assembly techniques that rely on homologous annealing of the terminal sequences. The qPCR-based approach presented here should facilitate the development of DNA assembly techniques and the diagnosis of inefficient assemblies.
Keywords
Assembly efficiency ; DNA assembly ; qPCR ; Secondary structure ; Transformation
SupplementaryMaterials
Figures
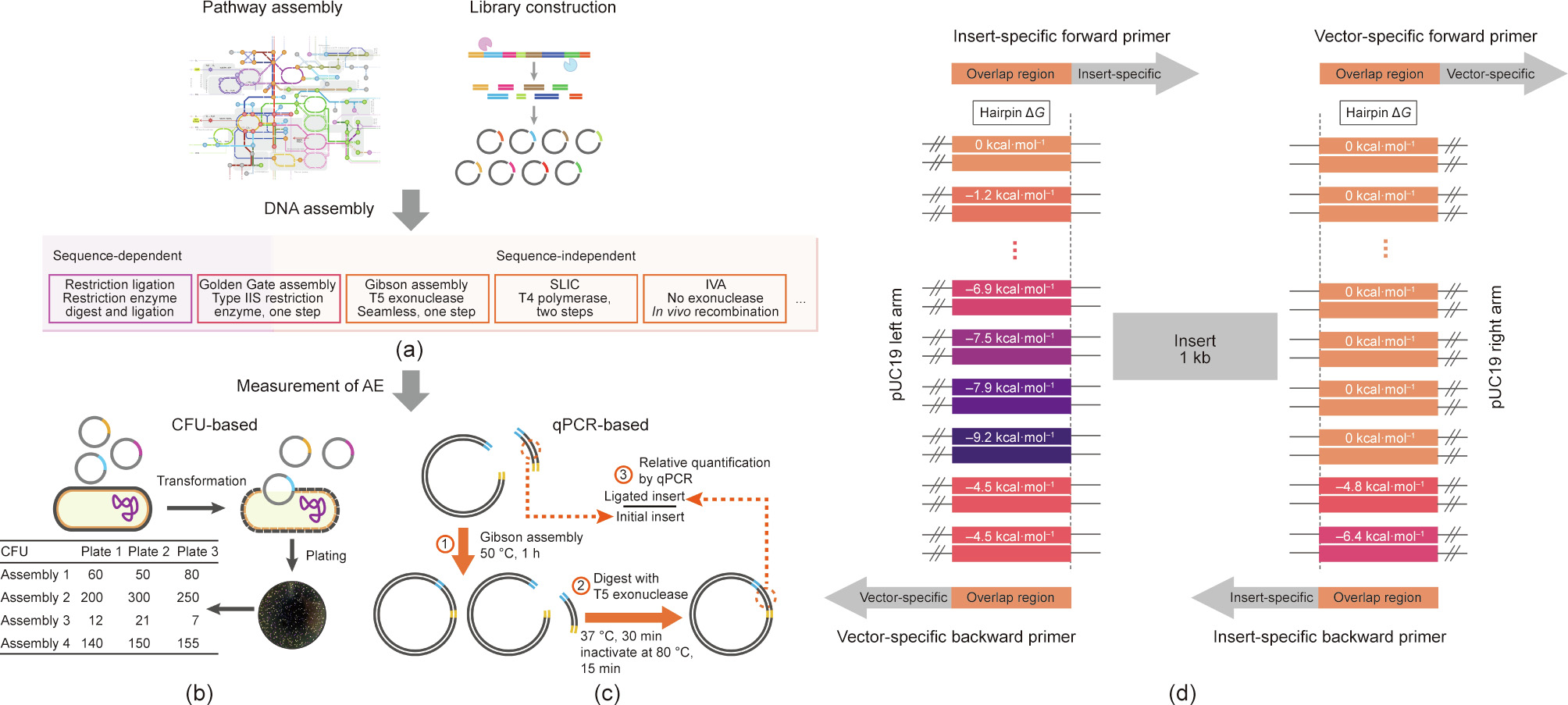
Fig. 1

Fig. 2

Fig. 3

Fig. 4

Fig. 5
References
[ 1 ] Andrianantoandro E, Basu S, Karig DK, Weiss R. Synthetic biology: new engineering rules for an emerging discipline. Mol Syst Biol 2006;2 (1):2006.0028. link1
[ 2 ] Choffnes ER, Relman DA, Pray L. The science and applications of synthetic and systems biology: workshop summary. Report. Washington, DC: National Academies Press; 2011. link1
[ 3 ] Ramon A, Smith HO. Single-step linker-based combinatorial assembly of promoter and gene cassettes for pathway engineering. Biotechnol Lett 2011;33 (3):549–55. link1
[ 4 ] Wingler LM, Cornish VW. Reiterative recombination for the in vivo assembly of libraries of multigene pathways. Proc Natl Acad Sci USA 2011;108 (37):15135–40. link1
[ 5 ] Dietrich JA, McKee AE, Keasling JD. High-throughput metabolic engineering: advances in small-molecule screening and selection. Annu Rev Biochem 2010;79(1):563–90. link1
[ 6 ] Purnick PEM, Weiss R. The second wave of synthetic biology: from modules to systems. Nat Rev Mol Cell Biol 2009;10(6):410–22. link1
[ 7 ] Nielsen AAK, Der BS, Shin J, Vaidyanathan P, Paralanov V, Strychalski EA, et al. Genetic circuit design automation. Science 2016;352(6281):aac7341. link1
[ 8 ] Chao R, Yuan Y, Zhao H. Recent advances in DNA assembly technologies. FEMS Yeast Res 2015;15(1):1–9. link1
[ 9 ] Quan J, Tian J. Circular polymerase extension cloning of complex gene libraries and pathways. PLoS One 2009;4(7):e6441. link1
[10] Li MZ, Elledge SJ. Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC. Nat Methods 2007;4(3):251–6. link1
[11] Cobb RE, Ning JC, Zhao H. DNA assembly techniques for next-generation combinatorial biosynthesis of natural products. J Ind Microbiol Biotechnol 2014;41(2):469–77. link1
[12] García-Nafría J, Watson JF, Greger IH. IVA cloning: a single-tube universal cloning system exploiting bacterial in vivo assembly. Sci Rep 2016;6(1):27459. link1
[13] Liang J, Liu Z, Low XZ, Ang EL, Zhao H. Twin-primer non-enzymatic DNA assembly: an efficient and accurate multi-part DNA assembly method. Nucleic Acids Res 2017;45(11):e94. link1
[14] Jin P, Ding W, Du G, Chen J, Kang Z. DATEL: a scarless and sequenceindependent DNA assembly method using thermostable exonucleases and ligase. ACS Synth Biol 2016;5(9):1028–32. link1
[15] Zhang Y, Werling U, Edelmann W. SLiCE: a novel bacterial cell extract-based DNA cloning method. Nucleic Acids Res 2012;40(8):e55. link1
[16] Engler C, Kandzia R, Marillonnet S. A one pot, one step, precision cloning method with high throughput capability. PLoS One 2008;3(11):e3647. link1
[17] Fu C, Donovan WP, Shikapwashya-Hasser O, Ye X, Cole RH. Hot fusion: an efficient method to clone multiple DNA fragments as well as inverted repeats without ligase. PLoS One 2014;9(12):e115318. link1
[18] Yoshida N, Sato M. Plasmid uptake by bacteria: a comparison of methods and efficiencies. Appl Microbiol Biotechnol 2009;83(5):791–8. link1
[19] Aune TEV, Aachmann FL. Methodologies to increase the transformation efficiencies and the range of bacteria that can be transformed. Appl Microbiol Biotechnol 2010;85(5):1301–13. link1
[20] Sleight SC, Bartley BA, Lieviant JA, Sauro HM. In-Fusion BioBrick assembly and re-engineering. Nucleic Acids Res 2010;38(8):2624–36. link1
[21] Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA 3rd, Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 2009;6(5):343–5. link1
[22] Livak KJ, Schmittgen TD. Analysis of relative gene expression data using realtime quantitative PCR and the 2-DDCT method. Methods 2001;25(4): 402–8. link1
[23] De’ath G. Boosted trees for ecological modeling and prediction. Ecology 2007;88(1):243–51. link1
[24] SantaLucia J Jr, Hicks D. The thermodynamics of DNA structural motifs. Annu Rev Biophys Biomol Struct 2004;33(1):415–40. link1
[25] Lomzov AA, Vorobjev YN, Pyshnyi DV. Evaluation of the Gibbs free energy changes and melting temperatures of DNA/DNA duplexes using hybridization enthalpy calculated by molecular dynamics simulation. J Phys Chem B 2015;119(49):15221–34. link1
[26] Gao Y, Wolf LK, Georgiadis RM. Secondary structure effects on DNA hybridization kinetics: a solution versus surface comparison. Nucleic Acids Res 2006;34(11):3370–7. link1
[27] Lu YP, Zhang C, Lv FX, Bie XM, Lu ZX. Study on the electro-transformation conditions of improving transformation efficiency for Bacillus subtilis. Lett Appl Microbiol 2012;55(1):9–14. link1
[28] Tee KL, Grinham J, Othusitse AM, González-Villanueva M, Johnson AO, Wong TS. An efficient transformation method for the bioplastic-producing ‘‘Knallgas” bacterium Ralstonia eutropha H16. Biotechnol J 2017;12(11):1700081. link1
[29] Russo R, Panangala VS, Wood RR, Klesius PH. Chemical and electroporated transformation of Edwardsiella ictaluri using three different plasmids. FEMS Microbiol Lett 2009;298(1):105–10. link1
[30] Bzymek M, Lovett ST. Instability of repetitive DNA sequences: the role of replication in multiple mechanisms. Proc Natl Acad Sci USA 2001;98 (15):8319–25. link1
[31] Lovett ST. Encoded errors: mutations and rearrangements mediated by misalignment at repetitive DNA sequences. Mol Microbiol 2004;52 (5):1243–53. link1
[32] Young CL, Britton ZT, Robinson AS. Recombinant protein expression and purification: a comprehensive review of affinity tags and microbial applications. Biotechnol J 2012;7(5):620–34. link1
[33] Pelicic V, Reyrat JM, Gicquel B. Expression of the Bacillus subtilis sacB gene confers sucrose sensitivity on mycobacteria. J Bacteriol 1996;178(4):1197–9. link1











 京公网安备 11010502051620号
京公网安备 11010502051620号




