

《1. Introduction》
1. Introduction
Transplantation is the optimal therapy for treating end-stage organ diseases, although transplant rejection remains a major limitation for long-term allograft survival. In general, after allografts are implanted, the innate immune cells start to mediate the innate immune response and inflammation; subsequently, the host adaptive response is activated to reject allogeneic organ grafts [1]. The limited effectiveness of improving long-term graft survival by current immunotherapeutic strategies that mainly, if not exclusively, target adaptive immune cells has prompted researchers to pay more attention to other immune cells beyond adaptive immune cells in mediating allograft rejection. In fact, innate immune cells such as dendritic cells (DCs), macrophages, and natural killer (NK) cells are closely involved in graft rejection, in addition to having inflammatory effects [2–4]. Moreover, there is compelling evidence that the role of innate immune cells in transplant immunity goes far beyond the traditional concept that innate immune cells participate in organ graft rejection predominantly by means of inflammation and their antigen-presenting ability [5–8], and it is believed that non-T cells, especially innate immune cells, are becoming more important in transplant responses [5,6].
Macrophages are highly heterogeneous and plastic cells whose phenotype and function are greatly regulated by their microenvironments [1,9–11]. Traditionally, naïve macrophages (M0) are functionally polarized into two subsets: M1 and M2 macrophages [12–16]. M1 is induced by interferon (IFN)- or lipopolysaccharide (LPS) + IFN-, and so forth, and its polarization relies on signal transducer and activator of transcription 1 (STAT1). M1 produces interleukin (IL)-1, IL-6, tumor necrosis factor (TNF)-α, and nitric oxide; it then associates with T helper 1 type (Th1) immune responses. M2 is induced by IL-4 and IL-13, and its polarization depends on STAT6. M2 produces IL-10, transforming growth factor (TGF)-β, and arginase. Functionally, M1 exhibits proinflammation and tissue injury capacities, whereas M2 has anti-inflammation, tissue repair, and pro-fibrosis functions. Moreover, it has been recently demonstrated that IL-23 can induce a unique subpopulation of macrophages mainly producing IL-17A, IL-17E, and IL-22 [17], and that M0 can also be polarized to haemoglobin/haptoglobin complexes-induced macrophages (HA-mac/M(Hb)), and heme-induced macrophages (Mhem) [18]. In addition, some macrophage subpopulations, such as regulatory macrophages (Mregs), exhibit the immunosuppressive function (Fig. 1).
or lipopolysaccharide (LPS) + IFN-, and so forth, and its polarization relies on signal transducer and activator of transcription 1 (STAT1). M1 produces interleukin (IL)-1, IL-6, tumor necrosis factor (TNF)-α, and nitric oxide; it then associates with T helper 1 type (Th1) immune responses. M2 is induced by IL-4 and IL-13, and its polarization depends on STAT6. M2 produces IL-10, transforming growth factor (TGF)-β, and arginase. Functionally, M1 exhibits proinflammation and tissue injury capacities, whereas M2 has anti-inflammation, tissue repair, and pro-fibrosis functions. Moreover, it has been recently demonstrated that IL-23 can induce a unique subpopulation of macrophages mainly producing IL-17A, IL-17E, and IL-22 [17], and that M0 can also be polarized to haemoglobin/haptoglobin complexes-induced macrophages (HA-mac/M(Hb)), and heme-induced macrophages (Mhem) [18]. In addition, some macrophage subpopulations, such as regulatory macrophages (Mregs), exhibit the immunosuppressive function (Fig. 1).
《Fig. 1》

Fig. 1. The roles of macrophage subsets in transplant immunity. M0 macrophages can be polarized into M1 and M2. M1 macrophages have a proinflammation effect, while M2 macrophages mainly have an anti-inflammation effect and promote vasculopathy and fibrosis. Allograft-infiltrated macrophages in acute rejection are mainly M1, while those in chronic rejection are M2. M0 can also be polarized to M17, HA-mac/M(Hb), and Mhem macrophages, although studies on their effects in transplant immunity are still limited. M0 can differentiate to Mregs, which exhibit an immunosuppressive function in transplant immunity.
Macrophages are the dominant infiltrated cell types in allografts during rejection episodes, and graft-infiltrated macrophages are closely associated with both the short- and long-term outcomes of organ transplantation [19–23]. Therefore, the role of macrophages in allo-immunity is very important, especially when the host adaptive response is inhibited. Einecke et al. [24] described the dynamic changes of macrophages in murine kidney allografts. These allografts exhibited increased macrophage activation-associated transcripts (including allograft inflammatory factor 1 (Aif1), nitric oxide synthase 2 (Nos2), and TNF-α) at one day post-transplantation and increased inner-graft macrophages from two days post-transplantation, followed by a progressive increase. Macrophages may contribute to transplant rejection through distinct pathways such as antigen processing and presentation, costimulatory signals, cytokine productions, cross-talk with other immune cells, and immune regulation. Importantly, we recently demonstrated that macrophages can directly mediate transplant organ or tissue graft rejection [8]. In addition, Mregs have been found to participate in immune tolerance to allografts in certain transplantation tolerance induction protocols [25–27]. In allo-immunity, macrophages may be the therapeutic target after organ transplantation [7]. In fact, research in murine cardiac allograft transplantation has shown that selectively inhibiting allograft-infiltrated macrophages using a nanotechnology approach efficiently promoted transplant tolerance [28,29]. In the present manuscript, we will discuss the distinctive roles macrophages play in organ transplantation, including the mechanisms of macrophages in graft rejection, the protective effects of immunosuppressive macrophages in organ graft acceptance, and the effect of immunosuppressive drugs on macrophages in transplantation settings.
《2. Accumulation of macrophages in allografts》
2. Accumulation of macrophages in allografts
Due to ischemia and reperfusion injury (IRI), inflammation, tissue damage, or other reactions, innate immune responses are inescapable after transplantation [30,31]. Many proteins are upregulated in allografts. In murine kidney allografts, IFN--inducible transcripts (including ubiquitin D (Ubd), C–X–C chemokine ligand 9 (Cxcl9), Cxcl10, and Cxcl11) were found to have progressively increased and macrophage activation-associated transcripts (including Aif1, Nos2, and TNF-α) to have significantly increased in allografts one day after transplantation [24]. Allograftproduced trafficking proteins have been shown to promote the graft infiltration of macrophages, including monocyte chemotactic peptide (MCP)-1; intercellular adhesion molecule (ICAM)-1; vascular cell adhesion molecule (VCAM)-1; macrophage inflammatory protein (MIP)-1α; regulated-on-activation, normal T cellexpressed and secreted chemokine (RANTES); and macrophage migration inhibitory factor (MIF) [32–37]. IRI has also been shown to promote inner-graft lipocalin-2 (Lcn2) expression, which enhanced macrophage infiltration into murine cardiac grafts [38].
Cellular interactions are also important for macrophage graft infiltration. It has been demonstrated that macrophage graft infiltration requires help from cluster of differentiation 4+ (CD4+ ) T cells [39], and it has even been hypothesized that macrophage graft infiltration is caused by T cell activation [40]. Moreover, the absence of B cells has been found to inhibit macrophage infiltration into rat kidney allografts [41]. In line with this interaction, macrophages were shown to increase dramatically in kidney allografts suffering acute rejection [42].
In addition to graft infiltration, macrophages present active proliferation in allografts [21,43,44]. Macrophage colony-stimulating factor (M-CSF) expression, which can promote the proliferation of macrophages, is significantly upregulated in allografts and in graft-infiltrated macrophages. In murine kidney allograft transplantation under acute rejection conditions, the M-CSF receptors c-FMS were restricted to graft-infiltrating macrophages, and antic-FMS antibody treatment significantly reduced the accumulation and local proliferation of macrophages in allografts [43,44]. Therefore, graft infiltration and inner-allograft proliferation contribute to the accumulation of macrophages in allografts, and targeting macrophage recruitment and proliferation might be an effective strategy for inhibiting allograft rejection.
《3. Macrophage and allograft rejection》
3. Macrophage and allograft rejection
《3.1. Effect of stimulatory signals of macrophages on host adaptive response》
3.1. Effect of stimulatory signals of macrophages on host adaptive response
As antigen-presenting cells, macrophages can stimulate host adaptive response through three signals: major histocompatibility complex (MHC)-peptide complex, co-stimulation, and cytokines. In regard to the role of macrophages in rejection, Jose et al. [43] summarized the potential mechanisms: First, macrophages serve as allo-antigen presenting cells; second, they produce proinflammatory cytokines to promote the inflammatory response; third, they release nitic oxide and reactive oxygen species (ROS) to take part in antibody-dependent and cell-mediated cytotoxicity; and fourth, they release profibrotic factors and matrix-metalloproteases to promote scar formation. Benichou et al. [30] also concluded that macrophages produce a wide variety of cytokines for cytotoxicity inflammation, including IFN-α/, TNF-α, IL-1, IL-6, IL-12, IL-18, IL-15, ROS, and granulocyte CSF (G-CSF).
Recently, the role of CD47 on macrophages has attracted a great deal of attention from the research community. Two decades ago, the signal regulatory protein α (SIRPα)–CD47 pathway was identified and was discovered to regulate macrophages by distinguishing self- and non-self-antigens and their phagocytosis [45]. CD47 is broadly expressed by virtually all cells and is termed a ‘‘don’t-ea t-me” signal, while SIRPα can be expressed by monocytes, macrophages, DCs, and granulocytes [46]. The role of SIRPα–CD47 interaction in cancer, autoimmunity, allotransplantation, and xenotransplantation has been identified. In allograft transplantation, CD47 expression on allografts may play distinct roles at different phases of transplantation.
First, CD47 expression on donor cells may benefit donor antigen-induced tolerance. Donor-specific transfusion (DST) before transplantation is useful to induce transplant tolerance, and CD47 expression on donor cells may be essential for allo-tolerance induced by DST. In murine skin transplantation, the absence of CD47 on donor cells result in a failure to prolong skin graft survival, and inhibit allo-responses [47,48]. This effect has also been shown in murine skin allograft transplantation following hepatocyte transplantation [49]. Zhang et al. [49] found that CD47-deficient allo-hepatocytes induced a more intense allo-response in the host in comparison with wild-type allo-hepatocytes. Mice with CD47- deficient allo-hepatocyte transplantation exhibited a higher capacity to reject the skin grafts in comparison with mice with wild-type allo-hepatocyte transplantation. In murine cardiac allograft transplantation, the elimination of CD47 on cardiac allografts induced the long-term survival of allografts in MHC-I and/or -II mismatched host mice [50].
Second, the expression of CD47 on allografts may improve IRI. In rat kidney transplantation, rat steatotic liver transplantation, and porcine cardiac allograft transplantation, CD47 blockade, which was used to perfuse grafts prior to transplantation, has been shown to reduce the IRI of allografts and improve graft function and survival [51–53].
Third, CD47 expression on allografts may be essential to the stable function and tolerance of allografts. In rat allogenic kidney transplantation, blocking SIRPα or CD47 with monoclonal antibody can break the established kidney allograft immune tolerance, as indicated by kidney allograft dysfunction and rejection episodes [54]. Nevertheless, the roles of macrophages and CD47 in transplant immunity need to be further determined.
《3.2. Macrophages and acute rejection》
3.2. Macrophages and acute rejection
In acute rejection, macrophages can serve as the antigenpresenting cells in adaptive responses (Fig. 2); however, the direct rejecting role of macrophages cannot be ignored. In the rat acute rejection model, deletion of macrophages was shown to potentially prolong cardiac allograft survival with a preserved function [55]. It has been reported that macrophages accounted for 80% of graftinfiltrated human leukocyte antigen (HLA)-DR+ cells in human allografts [21]. This finding means that allograft-infiltrated macrophages preserve strong antigen presentation ability. In addition, it has been found that the numbers of allograft-infiltrated macrophages correlate with the severity of T cell-mediated rejection [21,56]. Data from human percutaneous renal biopsies showed that macrophages accounted for 52% of the infiltrated cells in allografts suffering mild rejection and 60% of the infiltrated cells in allografts suffering severe rejection [56]. Moreover, allograftinfiltrated macrophages were skewed to M1 polarization in acute rejection [54,57,58]. Intravascular activation of macrophages is associated with acute vascular rejection, and those activated macrophages may contribute to myocyte necrosis by compromising blood flow in small vessels, as evidenced by biopsy specimens of heart transplant patients [59].
《Fig. 2》
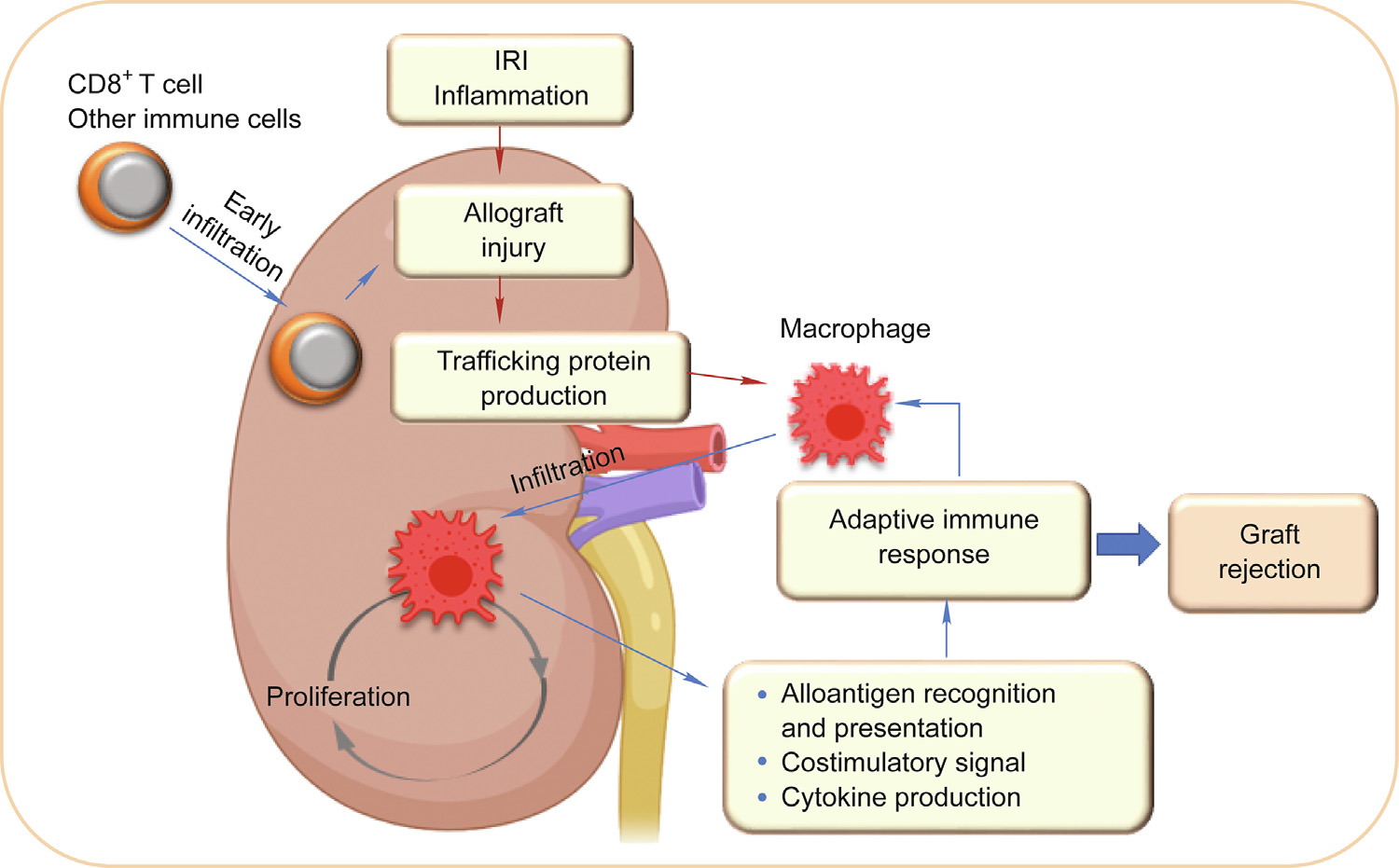
Fig. 2. Macrophages trigger and amplify the adaptive immune response to organ allo-grafts. Organ graft damage and other immune cells cause macrophage infiltration to allografts. The recruited macrophages can proliferate locally to increase the cell number. Macrophages can trigger and amplify the adaptive immune response through antigen presentation, costimulatory signals, and cytokine production.
《3.3. Macrophages and chronic rejection》
3.3. Macrophages and chronic rejection
Chronic rejection is the most important barrier to long-term allograft survival; unfortunately, treatment options for chronic rejection are still limited [60,61]. Vasculopathy and interstitial fibrosis are the major pathological hallmarks of chronic rejection, and macrophages play an important role in the development of these pathological changes. Macrophage depletion has been shown to significantly reduce murine cardiac allograft vasculopathy [62]. It has also been demonstrated that macrophage infiltration is associated with tubular injury and fibrosis progression [20]. Macrophages can promote fibrosis through two mechanisms: by promoting the accumulation, proliferation, and activation of fibroblasts; or by transiting into myofibroblasts [63]. M2 macrophages can produce TGF-β, which is the primary factor driving fibrosis [64]. In allografts suffering ongoing injury, macrophage– myofibroblast transition (MMT) has been observed. In human kidney allografts suffering chronic injury, M2 macrophages were co-located with α-smooth muscle actin (SMA)+ myofibroblasts in the areas of active interstitial fibrosis [65]; MMT was also obvious, characterized by a large amount of CD68+α-SMA+ MMT cells, predominantly with M2 polarization [66].
M2 polarization is predominant in graft-infiltrated macrophages in allografts suffering chronic rejection [20,39,60,65,67]. Human renal allograft biopsies taken one year after transplantation showed that 92% of graft-infiltrated macrophages had M2 polarization [20]. In murine cardiac allografts under chronic rejection conditions, M1 macrophage markers (IL-1β, IL-6, IL-15, IL-18, TNF-α, and Nos2) increased at two weeks but decreased by six weeks, whereas the M2 macrophage markers (a member of the chitinase family (Ym1), found in inflammatory zone 1 (Fizz1), vascular endothelial growth factor (VEGF), TGF-β, and CD206) increased by six weeks [39].
In terms of the role of macrophages in allograft vasculopathy and interstitial fibrosis, some regulatory pathways have been defined. Mammalian target of rapamycin (mTOR) and tumor necrosis factor receptor associated factor 6 (TRAF6) are pivotal in M1 and M2 polarization [6]. Deficiency of mTOR can impair M2 polarization, and then inhibit the chronic rejection of murine cardiac allografts, while TRAF6 deficiency can impair M1 polarization, and then induce severe vasculopathy in murine cardiac allografts under chronic rejection conditions. Furthermore, the mTOR signal plays an important role in programmed death ligand-1 (PD-L1). It has been found that mTOR deficiency upregulates the PD-L1 expression of macrophages in murine cardiac allografts, and PDL1 blockade restored the inhibited chronic rejection caused by mTOR deficiency in macrophages [6]. Ras homolog gene family member A (RhoA) deficiency inhibited macrophages from infiltrating into murine cardiac allografts by means of downregulated C– X3–C chemokine receptor 1 (CX3CR1), and then inhibited chronic rejection [68]. In murine cardiac allograft with chronic rejection, graft-infiltrated M2 macrophages expressed purinergic P2X7 receptor (P2X7R). P2X7R antagonist treatment has been found to inhibit the M2 induction and graft infiltration of macrophages, and then significantly alleviate allograft vasculopathy and prolong allograft survival [60]. In addition, MMT can be regulated by the small mother against decapentaplegic family member 3 (Smad3)- dependent mechanism in mouse kidney allografts [66].
Overall, macrophages promote the chronic degeneration of allografts mainly by means of M2 polarization. It seems that abolishing the effect of macrophages in allografts may effectively reduce allograft vasculopathy and interstitial fibrosis; however, more studies are needed to determine the mechanisms regulating macrophages and treatment targets in chronic rejection.
《3.4. Rejection and memory capacities of macrophages》
3.4. Rejection and memory capacities of macrophages
Traditionally, macrophages are mainly considered to propagate and amplify T and B cell-mediated adaptive immunity during allograft rejection. However, increasing evidence has demonstrated that macrophages can directly mediate allograft rejection (Fig. 3). Chu et al. [8] have demonstrated that macrophages can dominate acute rejection under certain conditions. In immunodeficient mice without T cells and B cells, adoptive transferred allogeneic antigen-primed macrophages (but not unprimed macrophages) were able to reject the skin allografts. We also found that the perforin pathway is involved in primed macrophage-mediated rejection. Other researchers have also consistently found that primed macrophages can mediate rejection through phagocytosis rather than direct killing [69]. In fact, graft-infiltrated macrophages present high phagocytic ability and protease activity [70], and allograft-induced macrophages present cytotoxic activity to allograft [71] or allo-tumor graft in a Ca2+-dependent manner [72].
《Fig. 3》

Fig. 3. Primed macrophages directly mediate allogeneic organ graft rejection. Macrophages activated in the presence of CD4+ T cells will gain the ability to directly reject allografts as effector cells. The primed macrophages may reject allografts by the perforin pathway, phagocytosis, or Ca2+-dependent pathways. CD40L: CD40 ligand.
Chu et al. [8] have shown that macrophages exhibit antigen specificity—namely, immunologic memory—in allograft rejection. In line with our results, a recent study also found that host monocyte/macrophages acquire specific memories after encountering allo-antigens [73]. This study also interestingly found that paired immunoglobulin-like receptors (PIR)-A are involved in the development of this antigen specificity. In addition, cellular help is useful for the generation of antigen-specific macrophages. Chu et al. [8] and Liu et al. [69] have found that macrophages require help from CD4+ T cells during the priming period and that the CD40/ CD40 ligand (CD40L) pathway may be involved in delivering this help.
Therefore, macrophages can directly mediate rejection and preserve immunologic memory capacity, and these functions go beyond the scope of innate immunity. Deciphering macrophage activation and functional pathways to reject allografts will help the research community develop novel therapeutic paradigms to prevent allograft rejection.
《4. Regulatory roles of macrophages and tolerance induction in transplantation》
4. Regulatory roles of macrophages and tolerance induction in transplantation
Transplant tolerance is the ultimate goal of organ transplantation. Macrophages may play a protective role in tolerance induction through their immunosuppressive subsets (Fig. 4). Researchers do not yet agree on the definition of immunosuppressive macrophage subsets due to the shortage of specific biomarkers and the lack of a unified criterion. Suppressive macrophages are induced from bone-marrow-derived monocytes with M-CSF but without activation/maturation stimuli. These suppressive macrophages express typical macrophage phenotypes, including F4/80, CD169, CD64, and low/intermediate levels of MHC-II, but not Ly6C and Ly6G. It is believed that these phenotype macrophages may be the precursors of Mregs [74]. Mregs are induced from bone-marrow-derived monocytes with M-CSF and are activated by IFN-. They are the mature phenotype of macrophages and express typical markers including CD11a, CD11b, CD68, F4/80, CD14, podoplanin, CD127, CD301, CD16/32, CD64, Sialoadhesin (CD169), macrophage scavenger receptor (CD204), dectin-1, MHC-II, and CD80. However, they express low or no levels of CD86, CD40, Toll-like receptor 2 (TLR2), TLR4, Ly6C, and Ly6G [75]. Hutchinson et al. [76] used CD14–/low-HLA-DR+ CD80–/low -CD86+ CD16– TLR2– CD163–/low to isolate induced Mregs. It has been reported that dehydrogenase/reductase 9 (DHRS9) is a stable marker for human Mregs and can be used to distinguish Mregs from a variety of human monocyte-derived tolerogenic antigenpresenting cells [77]. It should be noted that, due to the shortage of specific biomarkers and the lack of clear definitions of Mregs and CD11b+ Ly6C+ Ly6G– M-MDSCs (M-MDSCs: monocytic myeloid-derived suppressor cells), some reported M-MDSCs may also be Mregs [26,27,78,79]. Therefore, clarification and precise definitions with clear standards and specific biomarkers for these cell subpopulations are urgently needed.
Immunosuppressive macrophages are capable of inhibiting the alloreactive immune cells and inducing Tregs or other regulatory immune cells in the transplant response (Fig. 4). Mregs can inhibit T cell proliferation by the inducible NOS (iNOS) pathway in an allo-antigen-specific or cell-contact manner [75]. Human Mregs can induce allogenic IL-10-producing T-cell immunoglobin and immunoreceptor tyrosine-based inhibitory motif domain (TIGIT)+ Foxp3+ (Foxp3: forkhead box P3) Tregs by the indoleamine 2,3-dioxygenase, TGF-β, retinoic acid, Notch, and progestogen-associated endometrial protein pathways [80]. It should also be noted that allogeneic donor CD4+ CD25+ Tregs promote the differentiation of IL-10-producing M2 macrophages in recipient mice [81]. Thus, Mregs and T cells may form a regulatory loop to downregulate immune response in recipients. It has been reported that DC-specific ICAM-grabbing non-integrin (DCSIGN)+ immunosuppressive macrophages are crucial mediators of immunological tolerance in murine cardiac allograft transplantation with anti-CD40L-induced immune tolerance [82]. The identified DC-SIGN+ suppressive macrophages can inhibit CD8+ T cell proliferation and infiltration to grafts and promote the expansion and graft infiltration of Tregs [82]. Simultaneous DC-SIGN engagement by fucosylated ligands and TLR4 signaling is required for the production of IL-10 and is closely involved in this tolerance induction protocol with costimulatory blockade [82]. In addition, the roles of Mregs in mixed allogeneic chimeras should be recognized, because donor macrophages maturing in chimeric mice have been shown to gain the ability to induce the recipient effector CD4+ T cells differentiating into Foxp3+ Tregs. Furthermore, these macrophages have the ability to stimulate third-party T cell proliferation and immune response [83]. Therefore, macrophages with regulatory function may be widely involved in transplantation immune response through multiple pathways.
《Fig. 4》
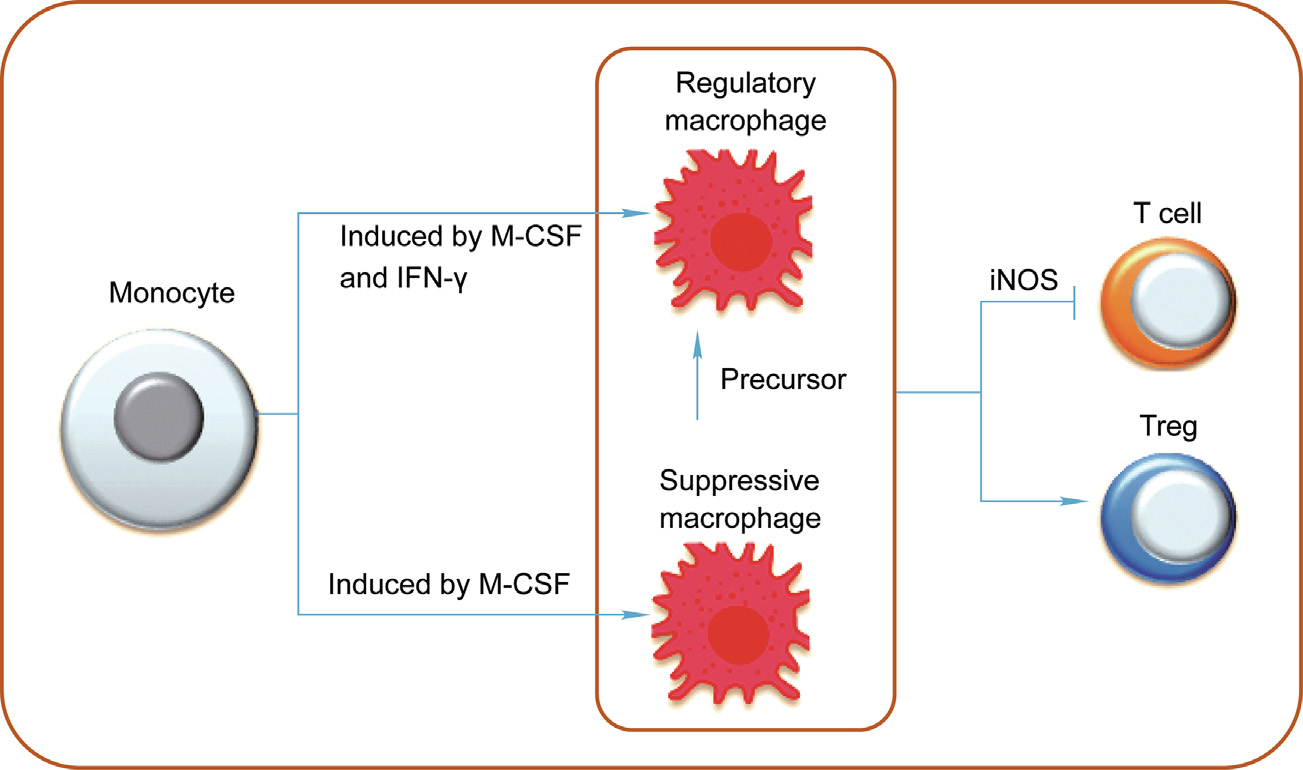
Fig. 4. The roles of immunosuppressive macrophages in allograft transplantation. Suppressive macrophages can be induced from monocytes by M-CSF, and regulatory macrophages can be induced from monocytes by M-CSF and IFN-. These immunosuppressive macrophages inhibit allograft rejection by suppressing effector immune cells and the induction of Treg cells.
The adoptive transfer of immunoregulatory cells as a means of establishing transplantation tolerance in clinical transplantation is now receiving significant attention and interest worldwide. Mregs may be a promising candidate cell type for avoiding allograft rejection and inducing immune tolerance, especially once an efficient and standardized manufacturing practice has been established [75,84]. The adoptive transfer of donor Mregs was shown to prolong mouse cardiac allograft survival, with synergy with shortterm rapamycin [75]. It was also demonstrated that the transferred Mregs survived for less than four weeks in mice, and that an infusion of 3 million, but not 1 million immunosuppressive macrophages—on the day before transplantation prolonged mouse skin allograft survival [74]. Human DHRS9+ Mregs, which are developed from M-CSF (CSF1)-stimulated CD14+ peripheral blood monocytes cultured with high concentrations of human serum [75,82,84–86], are currently being studied in the ONEmreg12 trial, a phase-I/II study of Mreg therapy, as a means of safely minimizing maintenance immunosuppression in kidney transplant recipients (clinicaltrials.gov: NCT02085629). Preliminary clinical research with two living-donor kidney transplant recipients indicated that the adoptive transfer of Mregs combined with low-dose tacrolimus treatment induced a good clinical outcome within 24 weeks [76]. The adoptive transfer of Mregs proved beneficial to one kidney recipient during a 7-year follow-up [80]. It is clear that more subsets of Mregs and the optimal protocols for tolerance induction with the adoptive transfer of Megs should be explored in the future.
《5. Effects of immunosuppressive drugs on macrophages》
5. Effects of immunosuppressive drugs on macrophages
Although there is increasing evidence of macrophages’ distinct roles in transplant immunity, our understanding of the effects of mainstream immunosuppressive drugs on macrophages is still limited. In the following section, we will briefly discuss the effects of clinical immunosuppressive drugs on macrophage function (Table 1 [6,21,65,87–99]).
《Table 1》
Table 1 The effect of immunosuppressive drugs on macrophages in transplantation.

《5.1. Effects of mTOR inhibitors on macrophages in transplantation》
5.1. Effects of mTOR inhibitors on macrophages in transplantation
The effect of mTOR inhibitors such as rapamycin on macrophages is better defined compared with those of other immunosuppressive drugs. Our previous results also demonstrated that mTOR deletion did not change the development of macrophages but only regulated monocyte/macrophage development in the early stages via the STAT5-interferon regulatory factor 8 (IRF8)- dependent CD115-expression pathway [87]. Rapamycin treatment did not inhibit the polarization of M1 macrophages but affected membrane phenotype and cytokine secretion in human macrophages. It significantly reduced the expression of CD25, TLR2, CD127, CD64, CD14, CD163, CD36, CD206, and CD209 but increased the expression of C–C chemokine receptor 7 (CCR7), CD86, and CD32 in M1 and reduced the expression of CD86, CD32, CD36, CD206, C–X–C chemokine receptor 4 (CXCR4), and CD209 in M2. Consistent with the finding that mTOR deficiency impairs M2 polarization [6], we found that rapamycin treatment negatively affected the survival of M2 macrophages [88].
Although conditional mTOR deficiency in macrophages does not affect the kinetics of acute rejection [6], Braza et al. [89] developed a rapamycin and high-density lipoprotein nano-immunotherapy that specifically targeted myeloid cells. This agent induced a longer survival of murine cardiac allografts by enhancing Mreg expansion. In the antibody-mediated rejection of mouse cardiac allograft transplantation, mTOR inhibitors significantly reduced macrophage infiltration into cardiac allografts by reducing ezrin/radixin/moesin (ERM) phosphorylation and ICAM-1 clustering in the MHC-I antibody-activated endothelium [99]. Aside from host macrophages, the adoptive transfer of donor Mregs before transplantation prolonged the survival of mouse cardiac allografts; this treatment was enhanced by rapamycin treatment [75]. Considering the inconsistency of the roles of mTOR in macrophages across different situations and models, further research on the effects of mTOR— including mTOR complex 1 (mTORC1) and mTORC2 in animal models and clinical samples—is required.
《5.2. Effects of calcineurin inhibitors on macrophages》
5.2. Effects of calcineurin inhibitors on macrophages
Clinical retrospective studies have demonstrated that tacrolimus is superior to cyclosporine A (CsA) in inhibiting the role of macrophages in chronic rejection. Clinical studies have consistently found that CsA has a superior effect on promoting interstitial fibrosis in kidney allografts in comparison with tacrolimus [90]. CsA may enhance the role of macrophages in chronic allograft injury [91]. First, CsA enhances the allograft infiltration of macrophages. It has been reported that CsA treatment promotes CCR5+ and CXCR3+ macrophage graft infiltration in rat kidney allograft transplantation [92]. In the kidneys of rats with CsA nephrotoxicity, significant macrophage infiltration was also observed [93]. Second, CsA may enhance the production of molecules in macrophages; these molecules are involved in chronic allograft injury, such as platelet-derived growth factor (PDGF) [94]. It has been inconsistently found that CsA inhibits tissue factor expression in LPS-induced macrophages by inhibiting the nuclear factor (NF)- jB pathway and inhibits the tissue factor associated with coronary lesion and fibrin deposition [95]. Interestingly, tacrolimus may not alter innate immune cell activity; however, it has been shown to preserve the anti-infection capacity of macrophages in mouse skin allograft transplantation [96]. It has also been demonstrated that tacrolimus does not disturb the phagocytosis of human monocytes to bacteria in vitro; only high concentrations of tacrolimus—not therapeutic concentrations—affect the maturation and polarization of macrophages, and the macrophage polarization is shifted to an M2-like phenotype in the presence of tacrolimus [97]. Thus, calcineurin has multiple effects on the function of macrophages in transplantation.
《5.3. Steroids to macrophages》
5.3. Steroids to macrophages
Steroids are also used extensively in clinical organ transplantation. In human allografts suffering antibody-mediated or T cell mediated rejection, the intensity of macrophage graft infiltration was found to be negatively associated with the effect of steroids[21]. In vitro, dexamethasone-treated human monocyte-derived macrophages expressed higher M2 markers and profibrotic cytokines, including TGF-β1, fibroblast growth factor (FGF)-2, and connective tissue growth factor (CTGF), but not M1-type cytokine TNF-α [65]. It was also observed that dexamethasone prompted the M2 polarization of rat macrophages in vitro [98]. In rats with induced mesangial proliferative glomerulonephritis, prednisolone treatment increased the glomerular M2 macrophages but had no effect on M1 macrophages [98]. Therefore, steroids may mainly enhance M2 polarization in transplantation settings.
《5.4. Effects of other immunosuppressive drugs on macrophages in transplantation》
5.4. Effects of other immunosuppressive drugs on macrophages in transplantation
To inhibit the effect of macrophages on rejection, other agents have been explored in animal and human studies. Researchers have demonstrated that mycophenolic acid (MPA) did not disturb the phagocytosis of human monocytes to bacteria in vitro but increased the expression of M2 surface markers on M1 macrophages [98]. Mizoribine treatment inhibited glomerular macrophage accumulation, selectively reduced M2 macrophages in vivo, and inhibited M2 polarization in vitro [99]. The erythropoietin (EPO) analogue ARA290 prevented early rat kidney allograft injury by targeting the NF-κB pathway and following reduced macrophage infiltration [34]. In allogeneic kidney transplant in rats with CsA treatment, simvastatin treatment inhibited macrophage graft infiltration and prevented allograft injury only under donor and recipient pretreatment plus daily recipient treatment [100]. In addition to inducing the long-term survival of murine cardiac allograft, anti-CD40L inhibited the graft accumulation and IFN- production of proinflammation macrophages [82]. A single-center clinical study with over 15 years of follow-up demonstrated that treatment with the macrophage inhibitor ibandronate was beneficial to human kidney allograft function and survival [101]. These immunosuppressive drugs may have potential applications in clinical transplantation after further study.
Further studies are needed to define the effect of current immunosuppressive agents, which mainly target adaptive immune cells, on macrophages. The results will contribute to the development of optimal immunosuppressive agents and drug application strategies.
《6. Conclusions and perspectives》
6. Conclusions and perspectives
Macrophages are the predominant immune cellular component accumulated in allografts, and may be used to predict allograft outcome. Macrophages not only trigger and amplify the adaptive immune response to donor antigens, but can also directly mediate allograft injury as effector cells. With their widely diversified function and plasticity, macrophages have many different subpopulations, including M1, M2, IL-23-induced macrophages (M(IL-23)), and Mregs. The different subpopulations of macrophages play distinctive roles in organ graft rejection. Approaches that target different subpopulations of macrophages, including inhibiting proinflammatory macrophages and promoting immunosuppressive macrophages, would offer novel therapies to avoid graft rejection and induce immune tolerance in clinical transplantation. In the future, more effort must be put into understanding the roles and molecular mechanisms of different subpopulations of macrophages in mediating allograft rejection or protection.
《Acknowledgments》
Acknowledgments
We would like to thank Mrs. Ling Li for her excellent laboratory management. This work was supported by grants from the National Key Research and Development Program of China (2017YFA0105002 and 2017YFA0104402 to Yong Zhao), the National Natural Science Foundation for General and Key Programs (C31930041 and C81530049 to Yong Zhao), Knowledge Innovation Program of the Chinese Academy of Sciences (XDA04020202-19 to Yong Zhao), the China Manned Space Flight Technology Project (TZ-1 to Yong Zhao), the National Natural Science Foundation of China (82070774), the Natural Science Foundation of Changsha (kq2007068), and the Natural Science Foundation of Hunan Province (2021JJ30965 and 2021JJ40866).
《Compliance with ethics guidelines》
Compliance with ethics guidelines
Liang Tan, Yinan Guo, Chang Feng, Yangxiao Hou, Xubiao Xie, and Yong Zhao declare that they have no conflict of interest or financial conflicts to disclose.











 京公网安备 11010502051620号
京公网安备 11010502051620号




