

《1. Introduction》
1. Introduction
Klebsiella pneumoniae (K. pneumonia) is ubiquitous in nature and infects a wide range of hosts, including plants, animals, and humans [1]. It is one of the leading inducements of clinical mastitis (CM) in dairy cows [2]—a prevalent and costly disease that is predominantly associated with bacterial infection [3,4]. In general, CM caused by Gram-negative bacteria is more difficult to cure than that associated with Gram-positive pathogens [5], with an average cost per case of 211.03 USD for Gram-negative bacterial infections compared with 133.73 USD for Gram-positive bacterial CM cases [6]. After Escherichia coli (E. coli), K. pneumoniae is the second most common Gram-negative cause of bovine CM, but it is the most detrimental in terms of decreased milk yield, discarded milk, treatment costs, death, and culling [7,8]. In view of the economic implications of K. pneumoniae infection in dairy farming, research into population structure, antibiotic resistance, and pathogenesis is particularly important.
On the basis of phylogenetic analyses using the sequences of gyrA, parC, and the chromosomally located β-lactamase gene, K. pneumoniae isolates can be classified into three distinct but closely related phylogroups: K. pneumoniae (KpI), Klebsiella quasipneumoniae (K. quasipneumoniae, KpII), and Klebsiella variicola (K. variicola, KpIII) [9–12]. All three phylogroups are associated with extraintestinal infections in humans, and reports from the United States have identified KpI, KpII, and KpIII isolates in milk samples from cows with mastitis [13,14]. However, because traditional laboratory diagnostic methods cannot reliably distinguish among the three phylogroups [15,16], estimating the true burden of each of the three Klebsiella phylogroups in bovine mastitis is still challenging.
To evaluate the impact of K. pneumoniae infection in dairy cows, most studies focus on virulence factors and antibiotic resistance. Several bacterial factors may contribute to K. pneumoniae infection in dairy cows, including the Fe3+ transport-associated fec genes, the lac operon, and genes related to metal (iron, zinc, and calcium) metabolism [14,17]. However, known pathogenicity factors that play a role in intestinal colonization and/or invasion in humans do not appear to be involved in the pathogenicity of K. pneumoniae in bovine CM. For example, clbA–Q (encoding colibactin), iucA–D and iutA (encoding aerobactin), irp, ybt, and fyu (encoding yersiniabactin), iroBCDEN (encoding salmochelin), mceA–J (encoding microcin), and rmpA and rmpA2 (regulator of mucoid phenotype A) are rarely observed in K. pneumoniae isolates recovered from cows with CM [17]. In addition, because K. pneumoniae responds poorly to antibiotic therapy, mastitis caused by this pathogen can result in significant economic losses [18].
Rates of antibiotic resistance among K. pneumoniae isolates from dairy cows vary significantly among regions. In Europe and the United States, Klebsiella spp. isolated from CM cases showed only low levels of resistance to tetracycline (5.6%–19.5%) and b-lactam antibiotics (0–6.9%) [19,20]. In China, a study revealed relatively high rates (10%–32%) of resistance to cefquinome, kanamycin, ceftiofur, polymyxin B, and tetracycline among Klebsiella spp [21]. K. pneumoniae isolates containing multiple antibiotic resistance genes, including those conferring resistance to β-lactams (blaCTX-M, blaSHV, and blaTEM), tetracyclines (tet(B)), and quinolones (oqxAB), have also been detected in cows from Europe and the United States [14,17,22,23]. Few studies, however, have examined the antimicrobial resistance profiles of K. pneumoniae isolates from cows with CM in China.
According to the China Dairy Statistical Yearbook 2017 [24], there were approximately 15 million head of dairy cows in China by the end of 2016, and the average economic loss associated with CM was 29–135 USD per cow per year [25]. Despite the importance and high resistance rates of K. pneumoniae in CM cases, little is known about the population structure and molecular characteristics of K. pneumoniae from cows with CM in China. This lack of information limits our understanding of the risks of K. pneumoniae to cows and hinders the identification of key control points. Herein, we continuously collected milk samples from cows with CM at three largescale dairy farms in Northern China from 2018 to 2019, detected the prevalence of K. pneumoniae isolates, and evaluated their antimicrobial susceptibility. We then used whole-genome sequencing (WGS) and bioinformatics analyses to systematically examine the population structure and molecular characteristics of CM-associated K. pneumoniae from cows in Northern China. Finally, we compared the whole-genome sequences of 100 KpI and 36 Klebsiella quasipneumoniae subsp. similipneumoniae (KpII-B) isolates from human clinical samples with the KpI and KpII-B sequences obtained in the current study in order to assess the relationships among bovine- and human-associated KpI and KpII-B strains at the genome level.
《2. Materials and methods》
2. Materials and methods
《2.1. Herds, sample collection, and bacteriological culture》
2.1. Herds, sample collection, and bacteriological culture
Samples were collected from January 2018 to December 2019 from three representative large commercial dairy farms located in Shandong, Hebei, and Heilongjiang provinces. Each dairy farm had 3000–5000 lactating Holstein-Friesian cows and was managed by the same corporate enterprise with similar feeding and management practices. Cows were fed total mixed rations, milked in a milking parlor, and housed in freestalls. Before milking, cows were screened for CM, and suspected cases were confirmed by the herd veterinarians based on visible symptoms including udder swelling, heat, hardness, redness, and/or milk presenting as watery with flakes, clots, or pus. The collection of milk samples and bacterial isolation were carried out as described previously [25,26]. In brief, milk samples were collected from individual quarters displaying obvious mastitis symptoms such as palpable inflammation of the udder (swelling, pain, and redness) and/or deterioration of milk secretion. All samples were stored at a low temperature (2– 8 °C) following collection. For each sample, 10 μL of milk was inoculated onto CHROMagar Orientation plates (CHROMagar Company, France) and incubated at 37 °C for 18–24 h. Suspected K. pneumoniae isolates (blue-colored colonies) were recovered and boiled to extract the DNA, which was used for 16S rRNA gene sequencing and matrix-assisted laser desorption/ionization timeof-flight mass spectrometry analyses with previously described primers [27].
《2.2. Antimicrobial susceptibility testing》
2.2. Antimicrobial susceptibility testing
Antimicrobial susceptibility testing of K. pneumoniae isolates was performed using the broth microdilution method according to the Clinical Laboratory and Standards Institute (CLSI) guidelines [28]. E. coli ATCC® 25922 was used for routine quality control (QC). All tested antibiotics are commonly used for human and/or animal infections, including ceftriaxone, ceftiofur, florfenicol, gentamicin, amoxicillin/clavulanate, kanamycin, ciprofloxacin, tigecycline, tri methoprim/sulfamethoxazole, tetracycline, meropenem, and polymyxin. The results were interpreted according to the CLSI documents VET08 [28] and M100-S28 [29], and the European Committee on Antimicrobial Susceptibility Testing (ECAST) guideline [30]. Values of MIC50 and MIC90 represent the minimum concentration at which an antimicrobial agent inhibits the growth of 50% and 90% of bacteria, respectively.
《2.3. Whole-genome sequencing》
2.3. Whole-genome sequencing
Genomic DNA was extracted from overnight cultures using a HiPure Bacterial DNA Kit (Magen, China). DNA libraries were prepared using a KAPA HyperPrep Kit (Roche, Switzerland), and 150 base pair (bp) paired-end sequencing was conducted using the Illumina HiSeq 2500 platform (Annoroad Genomics Co., China). Sequence reads were de novo assembled using SPAdes (version 3.13.0) with × 50 minimum assembly coverage [31]. A total of 100 human K. pneumoniae (KpI) and 36 human KpII-B WGS were downloaded from the National Center for Biotechnology Information (NCBI) database. The KpI sequences corresponded to human clinical isolates in China from 2016 to 2019, while the human KpII-B sequences corresponded to human isolates in China, Pakistan, Thailand, USA, Mexico, Greece, the Netherlands, and Nigeria from 2015 to 2020. Detailed strain information is provided in Table S1 in Appendix A.
《2.4. Molecular analysis》
2.4. Molecular analysis
K. pneumoniae phylogroups were determined using Kleborate (version 0.4.0) by comparing genome assemblies against a curated set of Klebsiella assemblies from the NCBI databases [17]. Known antibiotic resistance and virulence genes were identified using a read-mapping approach implemented in short read sequence typing for bacterial pathogens (SRST2) based on the K. pneumoniae bacterial isolate genome sequence database (BIGSdb) and virulence factor databases [32]. Multilocus sequence typing (MLST) was used to determine the sequence types (STs) of the K. pneumoniae isolates [33]. Simpson’s diversity index, calculated using BioNumerics (version 7.0;AppliedMaths, Belgium),was used to evaluate genotype diversity. A minimum spanning tree of all STs was generated using BioNumerics with the BURST algorithm [34]. Through the online website, the extended-spectrum β-lactamase (ESBL)-producing SHV variants were separated from the non-ESBL-producing ones [35].
《2.5. Single-nucleotide polymorphism filtering and phylogenetic analysis》
2.5. Single-nucleotide polymorphism filtering and phylogenetic analysis
All draft genomes were used for core-genome alignments, and single-nucleotide polymorphism (SNPs) were identified by mapping the core-genome sequences against the K. pneumoniae strain NTUH-K2044 reference genome [17]. A neighbor-joining (NJ) phylogenetic tree based on the multiple core-genome SNP alignments was constructed using Parsnp in the Harvest package (version 1.1.2) [36] and visualized using the Interactive Tree of Life (iTOL).
《2.6. Genome annotation and pan-genome analysis》
2.6. Genome annotation and pan-genome analysis
Assembled draft genomes were annotated using the rapid prokaryotic genome annotation tool Prokka [37]. The resulting general feature format version 3 (GFF3) files were used as the input file, and the pan-genome (including a gene_presence_absence.csv file) was generated using Roary [38] (version 3.11.2). Scoary, a genome-wide association study (GWAS) analysis software [39], was used to calculate the associations between the bacterial accessory genome and the host traits. Based on the results of the GWAS analysis and gene function annotation, we evaluated the factors that may affect the adaptability and virulence of K. pneumoniae in cows. Genes found in 5%–95% of K. pneumoniae genomes were defined as common accessory genes. Principal components analysis (PCA) of these common accessory genes was performed using the prcomp function in R (version 3.5.3) [17].
《3. Results》
3. Results
《3.1. Prevalence of K. pneumoniae in bovine CM milk samples》
3.1. Prevalence of K. pneumoniae in bovine CM milk samples
A total of 183 K. pneumoniae isolates were recovered in 6301 CM milk samples from the three dairy farms (Fig. S1 in Appendix A). The annual detection rate of K. pneumoniae was fairly consistent (p = 0.97) across the two sampling years, with a rate of 3.0% (92/3053, 95% confidence intervals (CI): 2.4%–3.7%) recorded in 2018 and a rate of 2.8% (91/3248, 95% CI: 2.2%–3.5%) in 2019. In addition, no significant difference was found in the annual detection rates among the three provinces. The highest annual prevalence of K. pneumoniae occurred at the farm in Hebei Province in 2018 (3.5%, 26/751, 95% CI: 2.3%–5.0%), while the lowest annual prevalence occurred in the herd from Heilongjiang Province in 2018 (2.5%, 21/831, 95% CI: 1.6%–3.8%) (Fig. 1 and Table S2 in Appendix A).
《Fig. 1》

Fig. 1. Annual detection rates of K. pneumoniae in milk samples from cows with CM in Northern China from 2018 to 2019.
《3.2. Population structure and genetic diversity of K. pneumoniae》
3.2. Population structure and genetic diversity of K. pneumoniae
WGS and analysis of the NJ phylogenetic tree generated from the allelic profiles of the core genomes revealed that the 183 K. pneumoniae isolates from cows with CM could be categorized into three distinct phylogroups: K. pneumoniae (KpI, 78.1%, 143/183), KpII-B, (20.2%, 37/183), and K. variicola (KpIII, 1.6%, 3/183) (Fig. 2 (a)). The average nucleotide sequence identity of the core genes between and within these phylogroups was 96.2%–96.6% and >99.4%, respectively (Table S3 in Appendix A). Further PCA analysis using 5174 common accessory genes present in 5%–95% of genomes confirmed that the 183 genomes could be separated into the three phylogroups (Fig. 2(b)), suggesting that all three phylogroups can cause bovine udder infection in China.
MLST analysis revealed significant genetic diversity within the three phylogroups. A total of 50 STs were observed among the 143 KpI isolates, with a Simpson’s diversity index score of 94.2%, while 26 STs were identified among the 37 KpII-B isolates, with a Simpson’s diversity index score of 97.9% (Table S4 in Appendix A). The three KpIII isolates belonged to two different STs, with a Simpson’s index score of 66.7% (Table S3). The most prevalent KpI genotypes (n ≥ 5 isolates) were ST2324 (18.9%, 27/143), ST107 (8.4%, 12/143), and ST116 (7.7%, 11/143), while not more than three of the KpII-B or KpIII isolates belonged to the same ST (Fig. 2(c)). Distribution and dynamics analysis of the KpI genotypes revealed no consistent predominant STs in cows with CM from the same herd. ST116 was the most prevalent ST in the herd from Heilongjiang in 2018 (42.9%, 9/21), while ST2324 (33.3%, 5/15) was the most prevalent in 2019 in that herd. ST2324 was the most prevalent ST in the herd from Shandong in 2018 (55.2%, 16/29), compared with ST107 (23.3%, 7/30) in that herd in 2019. However, no predominant KpI ST was observed in the herd from Hebei Province in both of the sampling years (Fig. 2(d)).
《Fig. 2》

Fig. 2. Population structure of the K. pneumoniae isolates. (a) Phylogenetic network and NJ phylogenetic analysis based on the allelic profiles of the core genes. (b) PCA analysis based on the presence of common (5%–95% prevalence) accessory genes in the 183 K. pneumoniae genomes. (c, d) Minimum spanning trees of the 183 K. pneumoniae isolates based on MLST analysis, revealing the distribution and dynamics of genotypes in cows with CM in Northern China.
《3.3. Antimicrobial susceptibility》
3.3. Antimicrobial susceptibility
Among the 183 K. pneumoniae isolates, high rates of resistance were observed for trimethoprim/sulfamethoxazole (97.3%, 178/183), while moderate rates of resistance were observed for tetracycline (20.2%, 37/183) and ceftiofur (14.8%, 27/183). In comparison, only low rates of resistance were observed for ceftriaxone (5.5%, 10/183), florfenicol (5.5%, 10/183), gentamicin (2.7%, 5/183), amoxicillin/clavulanate (1.1%, 2/183), kanamycin (1.1%, 2/183), ciprofloxacin (0.5%, 1/183), and tigecycline (0.5%, 1/183), and all isolates were sensitive to meropenem and polymyxin. However, differences in the resistance profiles of the isolates were observed among the three phylogroups. The distribution of the minimum inhibitory concentrations (MICs) of the 12 tested antibiotics against the KpI isolates was wider than that for the KpII-B and KpIII isolates, although similar values of MIC50 and MIC90 for all antibiotics except ceftriaxone, ceftiofur, and tetracycline were observed for all three phylogroups (Table 1).
《Table 1》
Table 1 Antibiotic resistance profiles of KpI (n = 143), KpII-B (n = 37), and KpIII (n = 3) isolates from cows with CM in Northern China over the 2018–2019 study period.

a Results were interpreted according to the CLSI documents VET08 and M100-S28 and the ECAST guidelines (2015).
《3.4. Antimicrobial resistance and virulence gene profiles》
3.4. Antimicrobial resistance and virulence gene profiles
In total, 57 antimicrobial resistance genes were detected among the 183 isolates (Table S5 in Appendix A), 78.9% (n = 45) of which were identified in KpI isolates. β-lactamase-encoding genes were the most abundant resistance genes, with 100% of the isolates carrying blaSHV (21.0% ESBL-producing blaSHV variants and 79.0% nonESBL-producing blaSHV variants), 3.5% carrying blaCTX-M (blaCTX-M-14 and blaCTX-M-15), and 2.8% containing blaTEM-1. Furthermore, the aminoglycoside resistance gene strAB, phenicol resistance genes catA and floR, sulfonamide resistance gene sul2, trimethoprim resistance gene dfrA, and tetracycline resistance genes tet(A) and tet(D) were detected in the KpI isolates, with prevalence rates ranging from 4.2% to 32.2% (Table S5 in Appendix A). In comparison, the KpII-B and KpIII isolates only harbored the β-lactam resistance genes blaOKP-B and blaLEN, respectively (Fig. 3(a)). We also observed that 27 KpI isolates, which were positive for the ESBL-encoding genes blaSHV-2a (n = 22), blaCTX-M-14 (n = 2), or blaCTX-M-15 (n = 3), exhibited high-level resistance to ceftiofur.
《Fig. 3》
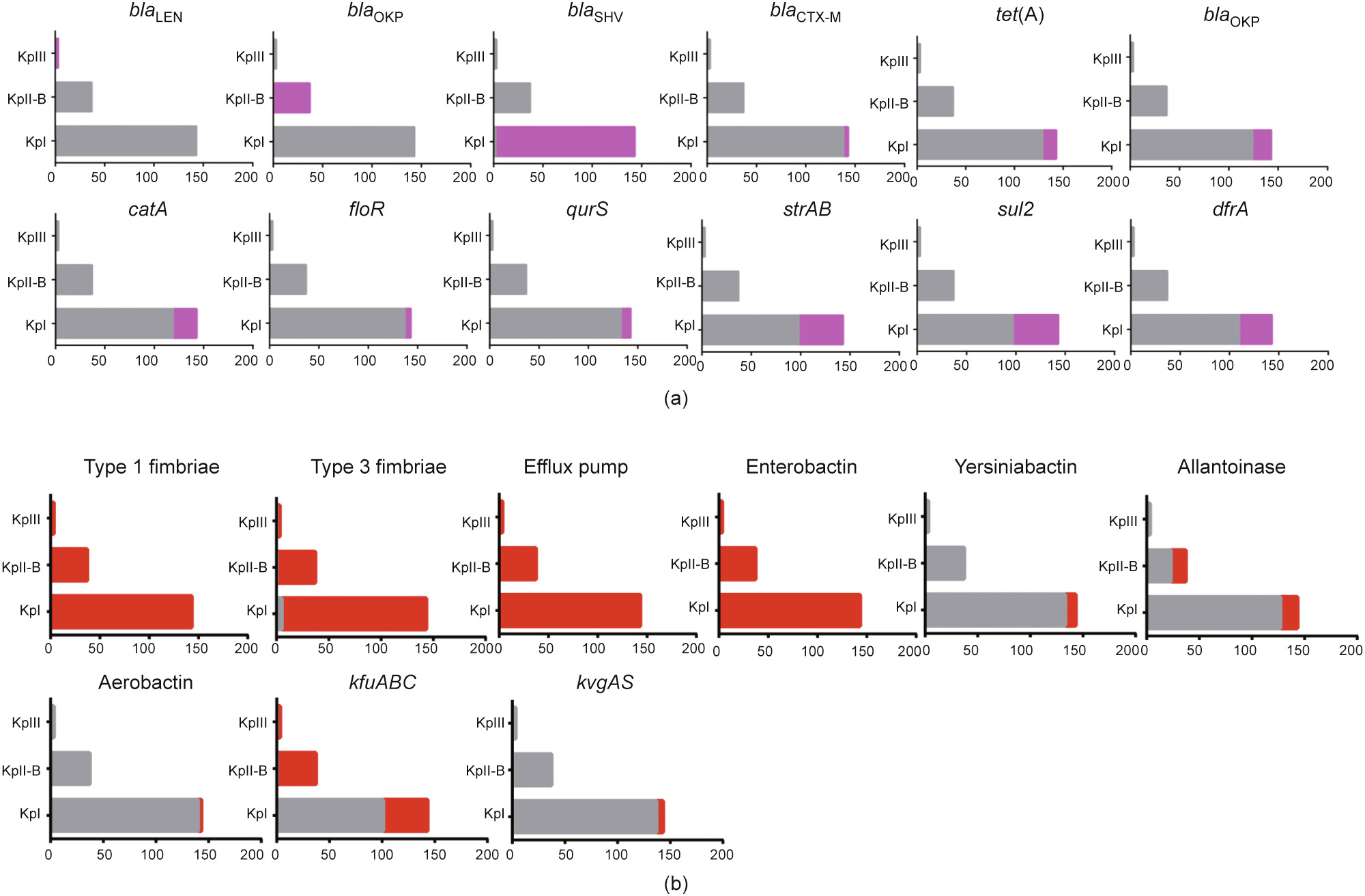
Fig. 3. Genomic characteristics of KpI (n = 143), KpII-B (n = 37), and KpIII (n = 3) isolates from cows. Bar plots show the presence (color) and absence (grey) of each of the (a) key resistance genes and (b) virulence factors within each lineage (KpI, KpII-B, and KpIII).
Overall, 70 virulence-associated genes were detected among the 183 isolates (Table S5). Genes coding for type 1 and type 3 fimbriae, which are major adhesive structures, and the AcrAB efflux pump, a novel virulence factor providing protection against the host innate immune system, were detected in all three phylogroups. However, the type 1 fimbrial regulation gene fimK was identified in all the KpI and KpIII isolates but was absent from the KpII-B isolates. Siderophore systems, including enterobactin, yersiniabactin, salmochelin, and aerobactin, are key virulence factors in K. pneumoniae and assist with the acquisition of iron—a limited resource—from the environment. Enterobactin-encoding genes were present in all the KpI, KpII-B, and KpIII isolates, while aerobactin (iucA–D and iutA; 2.1%) and yersiniabactin (irp, ybt, and fyu; 7.0%) genes were only detected in the KpI isolates. The two-component regulatory system encoding gene cluster kvgAS was also only found in KpI isolates (4.2%). Allantoin utilization genes (allABCDRS, ylbEF, glc, fdrA, and ybb) were identified in 37.8% of the KpII-B and 11.2% of the KpI isolates, while the ferric ion uptake operon kfuABC was found in all the KpII-B and KpIII isolates but in only 29.4% of the KpI isolates.
《3.5. Molecular characteristics and phylogeny of KpI isolates from cows and humans》
3.5. Molecular characteristics and phylogeny of KpI isolates from cows and humans
To determine the relationship between bovine and human K. pneumoniae isolates at the genome level and to evaluate the risk of bovine K. pneumoniae isolates to human health, we generated a NJ phylogenetic tree based on about 3 266 330 core-genome SNPs from 243 KpI genomes, including 100 publicly available KpI isolates from humans in China and the 143 isolates from the current study. The phylogenetic analysis revealed deep branching and complex and diverse population structures consistent with genotypes (Fig. 4(a)). Overall, the KpI isolates from cows showed greater genetic diversity than those from humans, with Simpson’s diversity indices for the MLST data of 94.2% and 40.4%, respectively. Although the strains from humans and cows did not form distinguishable host-specific clusters, obvious differences were found in the population structure between the human- and cowderived KpI isolates. ST2324, ST107, ST116, ST43, ST111, and ST2777 were common in cows with CM in Northern China, while ST11, ST23, and ST25 were frequently observed in Chinese human clinical isolates. Although ST11 was the predominant genotype (63%, 63/100) among the human KpI isolates, there was no consistently dominant genotype among the bovine isolates. Moreover, ST661, ST15, and ST37 KpI isolates were found in both humans and cows (Fig. 4(a)), and shared 2300–15 051 core-genome SNPs.
《Fig. 4》

Fig. 4. Comparative analysis of genomic characteristics of KpI isolates from human infections (n = 100) and cows with CM (n = 143). (a) NJ phylogenetic tree of KpI isolates based on core-genome SNPs. (b) PCA analysis based on accessory genes in the KpI genomes. (c) Detection rates for distinct genes in KpI isolates from cows and humans. *: p < 0.05: significant difference between KpI isolates from cows and humans.
Accessory genes among the 243 genomes were compared and analyzed, revealing 4432 common accessory genes present in 5%–95% of the genomes. Further PCA analysis showed that bovine- and human-derived KpI isolates cannot be reliably distinguished based on accessory gene analysis alone. However, there were significant differences in accessory genes between the human clinical ST11 isolates and the ST2324 isolates from cows with CM (Fig. 4(b)). In addition, nitrogen fixation-associated genes (nif operon) were detected in the ST2324 isolates, indicating that they may be derived from plants [17,40]. We then examined genes that were unique to KpI isolates from either cows or humans based on the results of GWAS analysis and the scanning of virulence genes. A total of 654 genes were identified as associated with cow-derived KpI (odds ratio (OR) > 1), and 1154 genes were found to be associated with human KpI (OR < 1) (Table S5). Among them, gene clusters associated with the synthesis of yersiniabactin (irp, ybt, and fyu), aerobactin (iucA–D and iutA), colibactin (clbA–Q), salmochelin (iroBCDEN), microcin (mceA–J), rmpA, and rmpA2 were significantly more prevalent (p < 0.01) in the human isolates than in the isolates recovered from cows. In comparison, clpC, lpfA, kfuABC, the lac operon genes (lacI, lacZ, and lacY), and Fe3+ transport proteinassociated genes (fecABDEIR) were more common in cow-derived isolates. In both cases, these unique genes may be beneficial for host invasion and adaptation, as well as evasion of the host immune response (Fig. 4(c)).
《3.6. Molecular characteristics and phylogeny of KpII-B isolates from cows and humans》
3.6. Molecular characteristics and phylogeny of KpII-B isolates from cows and humans
We next compared the phylogenetic characteristics of bovine and human KpII-B isolates. Because very few clinical KpII-B isolate genomes from China (n = 11) are available from the databases, we downloaded 25 genome sequences from Pakistan (n = 8), Thailand (n = 2), the United States (n = 5), Mexico (n = 1), Greece (n = 1), Netherlands (n = 1), and Nigeria (n = 7). Like the KpI isolates, the phylogenetic tree showed no hostspecific clusters for either the human or bovine KpII-B isolates (Fig. S2(a) in Appendix A). One human clinical KpII-B isolate (09A323) and two isolates from cows with CM (SD130-19 and SD52-19) showed a very close relationship, with 326 shared core-genome SNPs (326/3 296 574, 0.01% of the entire core genome). Human KpII-B isolate 09A323 was reported in Greece in 2019, while the two bovine KpII-B isolates (SD130-19 and SD52-19) were recovered from China in 2019. Although the human KpII-B isolate and the two bovine KpII-B isolates share a high degree of nucleotide sequence identity, which suggests that KpII-B may be transmitted between humans and cows, it is unclear how this transmission may occur.
We then examined virulence factors in KpII-B isolates from cows and humans. While the enterobactin synthesis operon clbA– Q and the two-component regulatory system gene cluster kvgAS was present in all the human and bovine KpII-B isolates, other known virulence factors were rarely detected. The prevalence of the allantoin utilization genes allABCDRS, ylbEF, glc, fdrA, and ybb was much higher in the bovine KpII-B isolates (37.8%) than in the human clinical isolates (13.9%). The prevalence of nif nitrogen fixation-associated genes was also high among the KpII-B isolates from both humans (44.4%) and cows (29.7%), indicating that these isolates may have originally been plant pathogens [17,40] (Fig. S2(b) in Appendix A).
《4. Discussion》
4. Discussion
In this work, we carried out a systematic analysis of K. pneumoniae isolates from cows with CM in China, focusing on prevalence, antimicrobial sensitivity, molecular characteristics, population structure, and the relationship between K. pneumoniae isolates from cows and those from humans. The annual prevalence (3.0% in 2018 and 2.8% in 2019) determined in our study was similar to the previously reported average prevalence (2.3%, 311/13 498) of Klebsiella spp. among seven Chinese provinces [25], but lower than that for Klebsiella spp. in herds in Northeast China (14.4%, 183/1271) [41]. For the first time, we classified three K. pneumoniae phylogroups—KpI, KpII-B, and KpIII—in cows with CM in China. These phylogroups have previously only been identified in cows with CM in the United States [14] and, consistent with our results, KpI was found to be the predominant phylogroup. However, no further analysis of the population structure, antimicrobial susceptibility profiles, or molecular characteristics of the phylogroups was conducted for the isolates from the United States. Similar to other studies of human clinical K. pneumoniae isolates [17,42], we identified high rates of KpI and low rates of KpII-B and KpIII in cows with CM in Northern China, as well as significant genetic diversity among the isolates.
Antimicrobial chemotherapy is commonly implemented for the prevention and control of bovine mastitis. However, with the increasing rates of antimicrobial resistance among mastitisassociated pathogens in dairy cows [43], this overuse may lead to the emergence of pan-resistant strains. In our study, the rates of resistance among KpI isolates to kanamycin (1.4%), amoxicillin/clavulanate (1.4%), and ceftiofur (18.9%) were lower than those reported for Klebsiella spp. isolated from cows with CM in large Chinese dairy herds in 2019 (15%, 38%, and 21%, respectively) [21]. The AmpC β-lactamase gene blaDHA and ESBL genes (blaCTX-M-14, blaCTX-M-15, blaSHV-27 and blaSHV-2a), which are usually associated with multidrug resistance of KpI isolates from humans [44], were respectively absent and relatively high (24.5%) in KpI from cows in this study. The low prevalence of blaCTX-M-14 (1.4%) and blaCTX-M-15 (2.1%) in the KpI isolates is consistent with the results for many different Enterobacteriaceae from cows [14,22,45], whereas blaSHV-2a, which was observed in a relatively high proportion (15.4%) of the KpI isolates in this study, has not previously been reported in cows. We then compared the virulence genes present in the isolates belonging to each of the three phylogroups. The ferric uptake operon kfuABC and allantoinase-related genes such as allABCDRS, ylbEF, glc, fdrA, and ybb were more prevalent in the KpII-B isolates than in the KpI isolates, while genes coding for aerobactin (iucA–D and iutA), yersiniabactin (irp, ybt, and fyu), and the KvgAS two-component regulatory system (kvgAS) were only detected in the KpI isolates. These findings are consistent with reports on human clinical K. pneumoniae isolates [17]. Importantly, we only found the type 1 fimbrial regulation gene fimK, which promotes K. pneumoniae virulence in murine pneumonia, in the KpI isolates [46]. Taken together, our results suggest that KpI isolates have greater pathogenic potential in cows than KpII-B and KpIII isolates based on the prevalence of antimicrobial resistance and virulenceassociated genes.
As well as being an important cause of mastitis in cows, KpI isolates are a common cause of human infections [47]. However, MLST analysis in our study showed significant differences in the population structure (genotypes) of the KpI isolates from cows and humans. ST11 and ST23 were the predominant genotypes in the human clinical isolates, with previous studies linking ST11 with multidrug resistance [48,49] and ST23 with hypervirulence [50,51]. In comparison, ST2324, ST107, ST116, ST43, ST111, and ST2777 isolates were more frequently observed in cows with CM. Genes associated with siderophore synthesis and capsular regulation were highly prevalent among the human clinical KpI isolates but were rare among the KpI isolates from cows. However, kfuABC was significantly more prevalent in the isolates from cows than in the human-derived isolates. These differences in population structure and virulence gene carriage may indicate that KpI isolates from cows pose relatively little threat to human health. Compared with the significant differences in population structure and pathogenic potential between the KpI isolates from cows and those from humans, the KpII-B isolates were more conserved. For example, cow- and human-derived KpII-B isolates shared branches on the phylogenetic tree and carried few known virulence genes. Plants like maize provide a suitable habitat for Klebsiella spp., which is capable of producing nitrogenase benefiting plant growth [40]. The nif genes were prevalent among all the KpII-B isolates, suggesting that they may have originally been plant commensal bacteria. In addition, two of the CM-derived KpII-B isolates shared a high level of nucleotide sequence identity with a human clinical KpIIB strain, raising the possibility of inter-species transmission.
We also found that clpC, lpfA, the lac operon genes, and Fe3+ transport protein-associated genes were more prevalent in bovine CM-associated KpI isolates than in human clinical isolates. These genes may therefore be important for the pathogenicity or host adaptability/specificity of K. pneumoniae in cows. Heat shock protein ClpC, a ClpATPase encoded by clpC, reportedly affects the intracellular survival capacity of Staphylococcus aureus in nonprofessional phagocytic cells [52]. LpfA, encoded by lpfA, is the major fimbrial subunit of long polar fimbriae, which is a key virulence factor in E. coli and aids in epithelial invasion during the establishment of mastitis [53–55]. Reports suggest that the lac operon genes (lacI, lacZ, and lacY) and Fe3+ transport proteinassociated genes (fecABDEIR) are essential for metabolism in KpI isolates, and may confer a selective growth advantage and adaptability in cows [14,17].
《5. Conclusions》
5. Conclusions
In summary, our study showed a low prevalence of K. pneumoniae in cows with CM in Northern China, but identified three phylogroups (KpI, KpII-B, and KpIII) among the isolates. KpI isolates are likely to be more harmful to cows than KpII-B and KpIII isolates, based on the prevalence of antimicrobial resistance and virulence genes. We conclude that KpI isolates from cows pose relatively little threat to human health due to differences in population structure and virulence gene carriage. Furthermore, the potential virulence factor-encoding genes kfuABC, clpC, and lpfA, the lac operon, and Fe3+ transport protein-associated genes, all of which were identified in the KpI isolates from cow mastitis, were rarely observed in the human clinical KpI isolates. Moreover, our results suggest that the KpII-B isolates may originate from plant pathogenic strains and are indicative of inter-host transmission between humans and cows, which should be monitored.
《Acknowledgments》
Acknowledgments
We thank the leaders and staff of the three dairy farms, especially Dr. Zunyang Zhao and Ms. Xia Zhang, for their friendly help in the sampling process. This study was supported by grants from the National Natural Science Foundation of China (81991535 and 81861138051), and the China Agriculture Research System (CARS-36).
《Compliance with ethics guidelines》
Compliance with ethics guidelines
Shikai Song, Wenjuan He, Dawei Yang, Manar Benmouffok, Yao Wang, Jiyun Li, Chengtao Sun, Xiangbin Song, Shizhen Ma, Chang Cai, Shuangyang Ding, Congming Wu, Zhangqi Shen, and Yang Wang declare that they have no conflict of interest or financial conflicts to disclose.
《Appendix A. Supplementary data》
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.eng.2021.01.015.











 京公网安备 11010502051620号
京公网安备 11010502051620号




