
《1. 引言》
1. 引言
对胎盘哺乳动物脊髓发育机制的研究在组织工程、神经科学和再生医学领域具有重要的意义[1]。在脊椎动物中,能够促进脊髓再生的内在生长能力在胚胎时期最明显[2‒3],在幼年阶段减弱,到了成年后几乎消失殆尽[4‒5]。到目前为止,从胚胎期到成年期脊髓发育过程中发生的转录变化,以及它们与脊髓内在生长能力的关系仍然尚不清楚。此外,也没有对发育中的大鼠脊髓进行长期基因表达模式系统性研究的报道。为了在组织水平上跟踪脊髓的长期发育过程,我们收集了大鼠胚胎第9天(E9d)至出生后第12周(P12w)的脊髓组织,进行RNA测序(RNA-seq)分析。从12个时间点共采集36个样本,来研究脊髓发育过程中相关基因表达的动力学过程。
《2. 材料与方法》
2. 材料与方法
《2.1. 大鼠脊髓样本的制备》
2.1. 大鼠脊髓样本的制备
实验所用的孕鼠、幼鼠和成年SPF级Sprague Dawley (SD)大鼠均由南通大学实验动物中心提供。取材时间为取材当日的上午6:30。大鼠被用乙醚吸入法麻醉,后置于冰块上用专用断头器(Pushin Zr-11; Shanghai Puxin Instrument Technology Co., Ltd., China)从颈部断头。尽快分离并取出脊柱,并用预先制备的氧饱和态的人工脑脊液(artificial cerebrospinal fluid, ACSF,冰水混合态溶液,成分:130 mmol∙L-1 NaCl, 5 mmol∙L-1 KCl, 2 mmol∙L-1 KH2PO4, 1.5 mmol∙L-1 CaCl2, 6 mmol∙L-1 MgSO4, 10 mmol∙L-1葡萄糖, 10 mmol∙L-1 HEPES;pH值为7.2,渗透压为305 mOsm)冲洗两次。然后在解剖显微镜(Olympus SZ51; Olympus Corporation, Japan)下,尽快在新鲜、冰水混合态的ACSF中解剖整个脊柱,并小心地剥去脊柱上的肌肉、骨骼、硬脊膜、蛛网膜、软脊膜和血管。采集从胚胎第9天(E9d)至出生后第12周(P12w)的大鼠脊髓组织样本,一共12个时间点(E9d、E11d、E14d、E18d、P1d、P3d、P1w、P2w、P3w、P4w、P8w和P12w),每个时间点3个生物重复。所有组织取出后立即在液氮中速冻,并保存在-180 °C的液氮中。
《2.2. RNA提取、文库制备和测序》
2.2. RNA提取、文库制备和测序
总RNA提取使用TRIzol试剂(Ambion, Inc., USA)按照制造商提供的说明操作。使用Epicentre Ribo-Zero RNA Removal Kit (Epicentre, USA)去除核糖体RNA (rRNA),并进行片段化和纯化,并使用Illumina HiSeq 2000系统(Illumina, Inc., USA)[6]进行测序。使用变性聚丙烯酰胺凝胶电泳(PAGE)方法分离小的非编码RNA(长度约18~30个核苷酸),该方法由Lagos-Quintana等[7]提出。然后使用TruSeq Small RNA Library Preparation Kit (Illumina, Inc., USA)来构建文库,并在Illumina HiSeq 2000平台上进行测序。
《2.3. RNA-Seq分析》
2.3. RNA-Seq分析
在开始分析前先对原始数据进行过滤,过滤条件如下:去除含接头(adapter)的读段;去除Ns比例高于10%的读段;去除低质量的读段(质量值Q ≤ 10的碱基数占整条读段的50%以上)。接下来,通过使用Short Oligonucleotide Alignment Program 2 (SOAP2) [8]将映射到rRNA序列的读段删除,以过滤掉rRNA污染物。大鼠参考基因组和注释文件(版本为Rn6.0)是从加利福尼亚大学圣克鲁兹分校(UCSC)基因组浏览器数据库(http://genome.ucsc.edu)中下载。为了量化基因表达,我们利用Bowtie2比对软件[9]将过滤后的reads比对到大鼠的参考基因集上,然后利用RSEM软件(RSEM是一种用于从RNA-Seq数据中估算基因和亚型表达水平的软件包)[10]分别计算参考基因表达水平和转录本表达水平。利用NOIseq [11]来识别两个连续发育期之间的差异表达基因(DEG)。采用FPKM (fragments per kilobase of exon per million mapped reads)标准方法对测序深度和基因长度进行归一化。将两个样本之间具有差异倍数log2的绝对值大于或等于1并且变异概率大于或等于0.8的基因定义为DEG。
《2.4. 阶段特异表达基因筛选》
2.4. 阶段特异表达基因筛选
为了解脊髓发育不同时期特异性表达的基因,我们采用组织特异性指数(tissue specificity index τ)来衡量不同时间点特异表达的基因[12]。τ的计算如公式(1)所示:
(1)
τ取值范围在0~1之间。τ接近0,表示该基因在所有样本中都有表达;τ接近1,表示该基因为某一时期特异性表达。本研究中将基因τ ≥ 0.9定义为时期特异性表达的基因。
《2.5. 加权基因共表达网络分析》
2.5. 加权基因共表达网络分析
为了识别脊髓发育过程中的遗传表达模式,我们进行了加权基因共表达网络分析。加权基因共表达网络分析(WGCNA)包[13]使用所有在E9d至P12w的样品中均有表达的基因的归一化表达数据(FPKM)。采用软阈值以确定合适的加权参数。再将模块内的基因列表上传到STRING数据库(https://string-db.org/)[14],获取模块内基因互作信息。利用Gephi软件[15]对整合了蛋白质相互作用(PPI)数据的网络模块进行可视化和分析。利用R软件包pheatmap对模块内的基因表达进行聚类分析和可视化。
《2.6. 基因功能富集分析》
2.6. 基因功能富集分析
利用Database for annotation, visualization and integrated discovery (DAVID)在线数据库[16‒17](http://david.abcc.ncifcrf.gov/)对基因进行功能富集分析。
《2.7. 可变剪接分析》
2.7. 可变剪接分析
使用TopHat [18]识别剪接点,并使用转录本剪接的重复多变量分析(rMATS)[19]检测两个相邻样本之间的差异可变剪接事件(DASE),如E9d vs E11d, E11d vs E14d, E14d vs E18d等。rMATS计算内含子和跳跃子类型的相对丰度,同时计算差异剪接的P值和错误发现率(FDR),差异剪接一般定义为FDR ≤ 0.05。
《2.8. 长链非编码RNA分析》
2.8. 长链非编码RNA分析
为了识别已知的和新的长非编码RNA(lncRNA),每个样本的RNA-Seq reads首先使用TopHat映射到基因组,然后用Cufflinks [20]组装。利用Cufflinks中的Cuffmerge和Cuffcompare对所有样本的转录本进行合并,并与已知的信使RNA(mRNA)和lncRNA进行比较。保留标记为“i”“j”“u”“x”和“o”的转录本。利用Coding Potential Calculator (CPC) [21]和iSeeRNA [22]预测新的转录本潜在编码能力。LncRNA的定量和差异表达分析与前文中RNA-Seq的分析相同(见2.3节)。
《2.9. 小RNA分析》
2.9. 小RNA分析
首先删除接头序列和低质量的读段。在去除掉rRNA、scRNA、snoRNA、snRNA和tRNA相关的读段后,将干净的序列在数据库中进行映射。剩下的序列分别使用Bowtie2和cmsearch [23]在miRbase (v21)和Rfam (v11)数据库中进行映射,以注释和分类小RNA [包括microRNA (miRNA)和Piwi-interacting RNA (piRNA)]。miRNA表达水平使用transcripts per kilobase million (TPM) [24]计算。用DEGseq [25]检测差异表达的小RNA。具有差异倍数log2的绝对值大于或等于1并且Q ≤ 0.001的miRNA被定义为显著差异表达。采用Ingenuity pathway analysis (IPA)来识别和提供潜在的miRNA的上游调控因子。
《2.10. 实时定量聚合酶链反应》
2.10. 实时定量聚合酶链反应
先使用TRIzol试剂分离总RNA,然后使用PrimeScript RT Reagent Kit (TaKaRa Bio, China)进行逆转录。基因表达定量采用SYBR Premix Ex Taq (Tli RNaseH Plus; TaKaRa Bio, China)的StepOnePlus RT-qPCR系统(Applied Biosystems, USA)。采用2-∆∆T法测定mRNA的相对表达量,采用GAPDH作为内参。所有实验均重复三次。
《2.11. 免疫荧光染色》
2.11. 免疫荧光染色
将大鼠脊髓组织用4%的多聚甲醛在室温下固定30 min后,4 ℃孵育过夜。用磷酸盐缓冲液(PBS, 0.01 mol∙L-1, 25 ℃, 21-040-CVR, Corning Inc., USA)洗三次(每次5 min)后,用免疫染色阻断液(Beyotime, China)阻断切片(1 h, 25 ℃)。切片与一抗于4 ℃下孵育12~16 h。PBS冲洗三次后,加入二抗,于25 ℃下孵育1 h,然后用Hoechst 33342染色15 min,每组重复三次。使用的抗体信息如下:anti-Dnmt3a (1:1000;ab188470; Abcam, UK), anti-NEUROD4 (1∶100; 14610-1-AP; Proteintech, USA), anti-Nestin (1∶200; MAB353; Merck Millipore, USA), anti-NeuN (1∶200; MAB377; Merck Millipore, USA), Cy3sheep anti-rabbit immunoglobulin G (IgG) (1∶500; C2306; Sigma-Aldrich, USA), Alexa Fluor 488 donkey anti-mouse IgG (1∶500; A21202; Invitrogen, USA)。
《3. 结果与讨论》
3. 结果与讨论
《3.1. 大鼠脊髓转录组数据概览》
3.1. 大鼠脊髓转录组数据概览
我们从转录组中鉴定了15 646个mRNA和667个miRNA。随后,我们随机筛选了两个基因和两个miRNA(Dnmt3, Neurod4, miR-29c-3p, miR-18a-3p),采用RT-qPCR实验和免疫荧光实验(IF)进行了验证[图1(a)、(b)]。我们还将我们的IF数据与存放在Allen Brain Atlas (ABA)数据库[26]中的小鼠脊髓原位杂交(ISH)数据进行了比较。总的来说,我们的IF结果与ISH数据一致[图1(a)中用星号标记],特别是出生后的样本是一致的(因为ABA数据库中只有出生后第4天和第56天两个点可参考)。这些结果证实了我们数据的质量,表明这些数据适用于下游的进一步分析。
《图1》
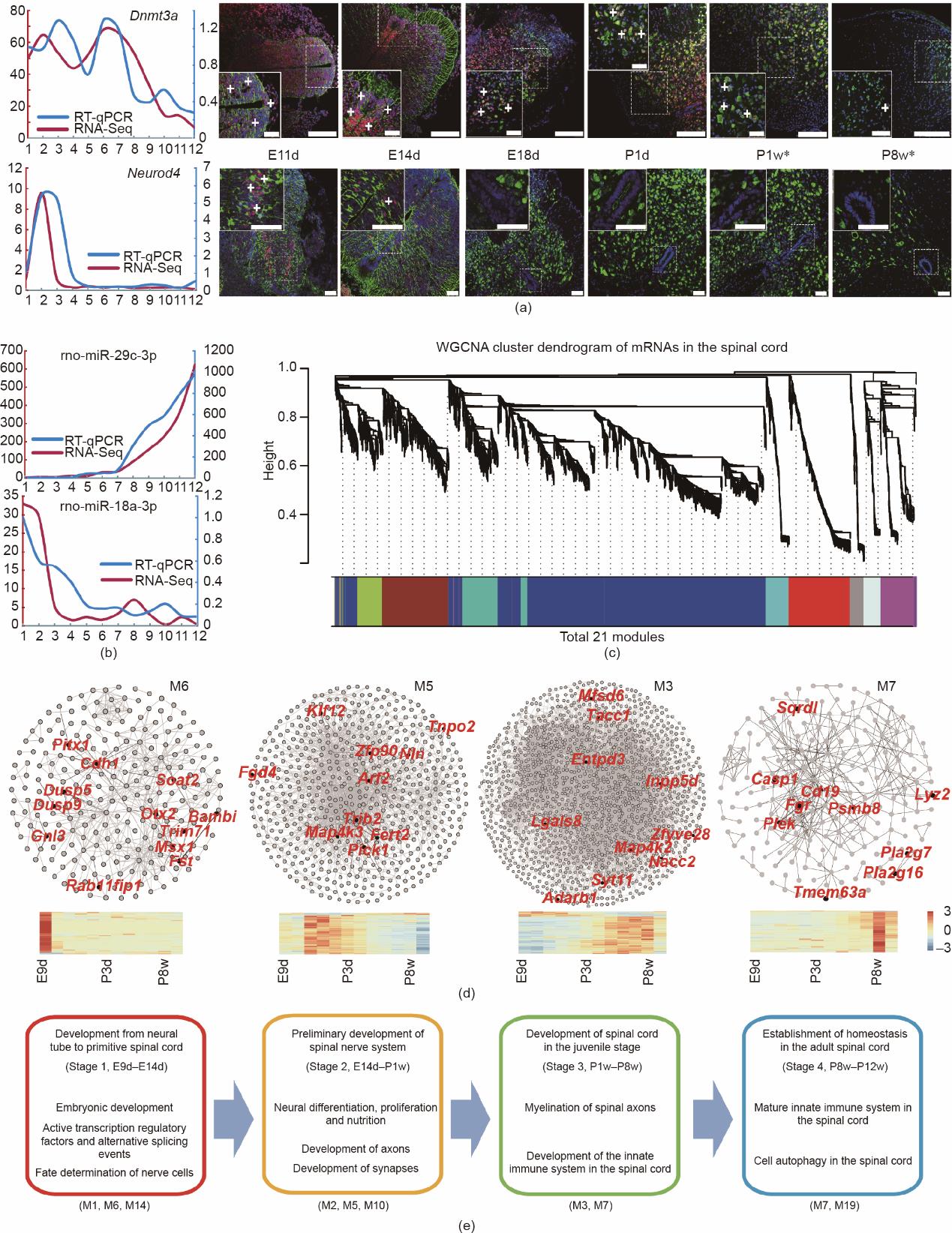
图1 大鼠脊髓发育过程中的重要基因表达验证和共表达网络的建立。
由于基因在参与相似的生物学通路或受相似的调控通路调控时会在宏观上展现出共表达的特征,共表达基因通过特定的算法分析后容易被聚集在一起[27]。加权共表达基因网络分析算法(WGCNA)是基于软阈值和无尺度网络,可以确定基因之间的相对关系,使得我们对共同表达基因模块的建立和相关生物学功能的研究成为可能[13,28]。应用WGCNA对生物功能模块进行研究,共鉴定出21个模块[M1~M21;图1(c)]。根据生物过程分析,这些模块的功能富集确定了脊髓发育的四个不同阶段[图1(c)]。第一阶段,即从E9d到E14d,由神经管形成原始脊髓。我们发现模块M6参与胚胎发育和基因转录调控,而模块M14则参与细胞命运的决定(见附录A中的表S1、表S2)。第二阶段(从E14d到P1w)是脊髓组织中神经系统的建立与发育时期,具体反映在与神经元的分化、增殖、营养、神经元投射发育、突触传递和轴突引导等相关生物学功能在这个阶段得到显著富集[见图1(e)、附录A中的表S3至表S5]。第三阶段(从P1w到P8w)的特征是参与髓鞘形成、先天免疫系统发育和氧化还原过程。而在第四阶段(从P8w到P12w)中先天免疫和细胞自噬相关基因被显著富集(见附录A中的表S6、表S7)。这些阶段的变化与相应时期[29]通过实验观察到的发育特征相互呼应。
此外,我们使用基因表达聚类分析来研究全部基因的表达模式,并确定了两种基本的表达模式[图2(a)中的Patterns 1和2]。Pattern 1的基因高表达在胚胎和新生儿期(共有7481个基因),其在胚胎发育中起着核心作用(见附录A中的表S8)。Pattern 2中的高度表达基因从幼年期持续到成年(共有6926个基因),主要参与能量代谢、免疫系统发育和细胞增殖(见附录A中的表S9)。
《图2》
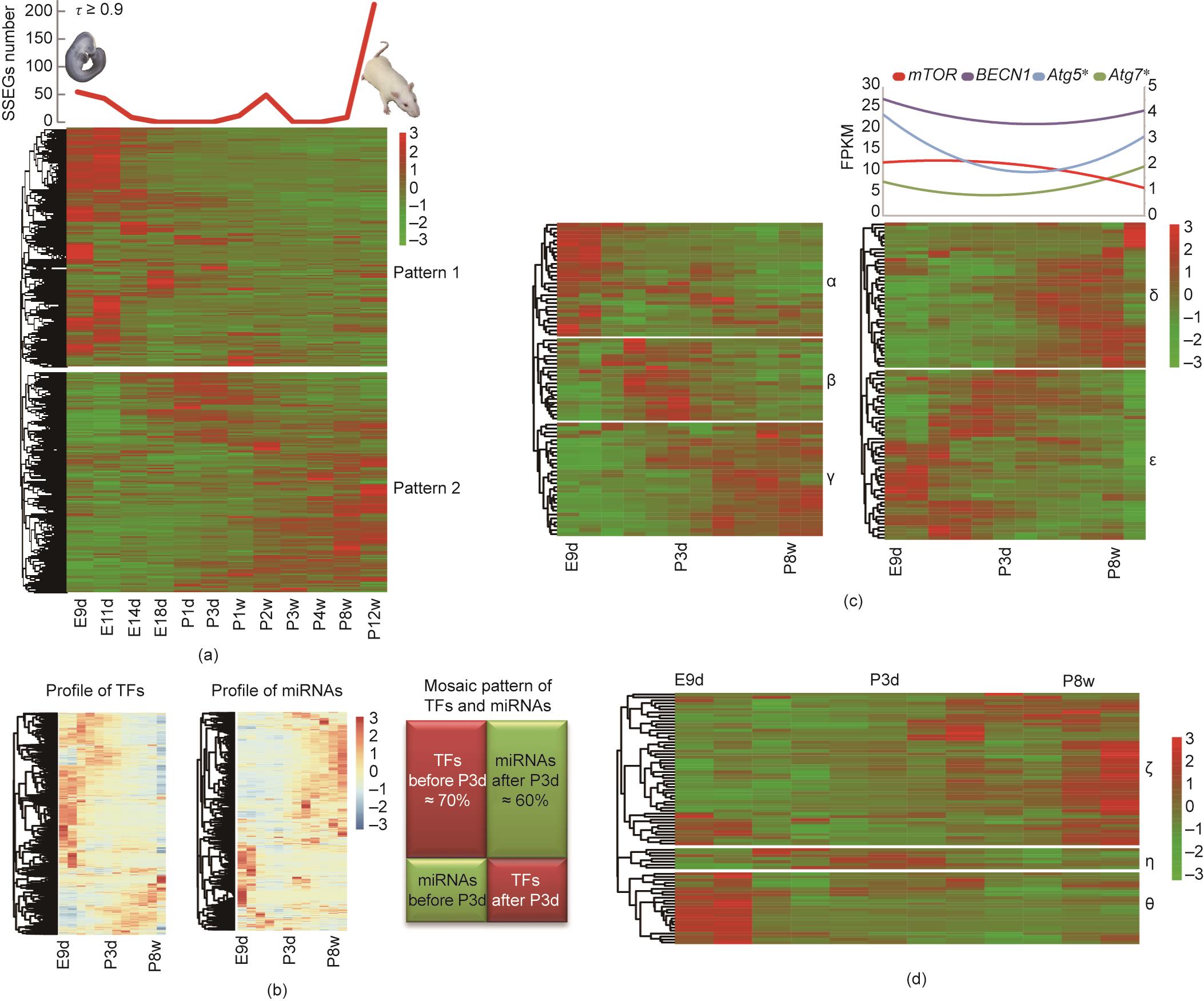
图2 大鼠脊髓发育过程中转录组基因、TF和miRNA的典型表达模式。
SSEG的分析有助于进一步了解与特定发育阶段相关的生物学过程[12]。一共有55个在E9d特异表达的SSEG,这些基因与上皮细胞分化、前后模式形成、神经板形态、胚胎发育和转录调控有关[见图2(a)、附录A中的表S10]。在E11d,共鉴定出43个与转录调控、神经元生成和组织稳态相关的SSEG。而在E14d一共有9个与蛋白质自身磷酸化、轴突、质膜组分和ATP结合相关的SSEG。有趣的是,在P8w和P12w中发现的SSEG都与免疫系统相关,如先天免疫、抗原处理和呈递(见附录A中的表S10)。
我们发现miRNA的表达模式在出生前后呈现两种趋势[miRNA的表达谱见图2(b)]。我们还观察到两种转录因子(TF)相关的表达模式,也是以新生期(P3d)为界限[TF的表达谱见图2(b)]。在P3d之前,与命运决定、干细胞功能调节和神经发育相关的TF被富集(见附录A中的表S11)。在P3d之后,与细胞分化和先天免疫激活相关的TF相对高表达(见附录A中的表S11)。我们还比较了miRNA和TF之间关系模式。并注意到,在P3d之前,大多数TF(约70%)的表达水平相对较高,而大多数miRNA(约60%)的表达水平相对较低;而P3d后,这种模式发生了逆转,TF的值约为30%,miRNA的值约为40% [镶嵌模式,如图2(b)所示]。这一发现提示一些调控系统(TFs-miRNA)在脊髓发育过程中可能具有阶段性协调作用。我们还注意到,大多数参与干细胞种群维持的基因可能在E9d和E11d活跃(见附录A中的图S1)。而对多巴胺能神经元和胆碱能神经元分化相关基因的分析表明,以P3d为时间基线,参与多巴胺能突触传递的基因比参与胆碱能突触传递的基因表达上调得更早(见附录A中的图S2),提示脊髓发育过程中功能性的多巴胺能神经元可能出现得相对更早。
为了研究脊髓发育过程中的能量和物质代谢,我们重点研究了能量储备代谢过程(energy reserve metabolic process)、自噬(autophagy)和氧化磷酸化(oxidative phosphorylation)等生物学过程中相关基因的表达模式[见图2(c)、(d),附录A中的表S12、表S13]。在能量储备代谢过程中[图2(c)的左侧],来自α和β分支簇的基因群富集于“正调控糖原生物合成过程”(positive regulation of glycogen biosynthetic process, P < 0.01;表S12)。来自β和γ分支簇的基因群富集于“cAMP生物合成的正向调控”(positive regulation of cAMP biosynthetic process, P < 0.01;见附录A中的表S12),由于ATP在腺苷酸环化酶的催化下发生环化形成cAMP,该通路的活跃提示成年期ATP合成的活跃。在新生期(P3d)前参与“自噬过程正调控”的相关基因群的聚类分析中[图2(c)的右侧],ε簇的基因群显著富集在“细胞对葡萄糖饥饿过程的反应”(cellular response to glucose starvation, P < 0.01;表S13)与“线粒体自噬的正调控”(positive regulation of mitophagy, P < 0.01;表S13);而在P3d后,δ簇显著富集于“异体吞噬的正调控”相关的基因显著富集(positive regulation of xenophagy, P <0.01;表S13),这种选择性自噬过程可能有回收能量和维持组织内稳态的功能,同时也是先天免疫的重要组成部分。有趣的是,对氧化磷酸化电子传递链中相关基因群的分析显示,大部分相关基因在向成年期脊髓发育的过程中逐渐高表达[block ζ;见图2(d)、附录A中的表S14],这种模式可能有助于为成年个体提供更充沛的能量来源。
《3.2. RNA表达的波动》
3.2. RNA表达的波动
通过比较相邻时间点(如E9d vs E11d, E11d vs E14d等)的DEG数量分布。我们在E11d vs E14d组鉴定到了2438个DEG [见图3(a)、附录A中的图S3和表S15]。从数字上看,这些DEG约占所有蛋白质编码基因的10%,也是其他组数量的近10倍。其中,三分之二的DEG上调并参与突触组装、轴突发生和神经系统发育等生物学功能[图3(b)中的红色柱]。其余下调的DEG参与了细胞分裂和细胞周期等生物学功能[图3(b)中的蓝绿色柱]。同时,差异表达的lncRNA和mRNA在这两个相邻时间点之间的分布也显示出相似的模式[图3(a)中的红色柱]。因此,我们把这种现象称为“RNA表达的波动”。为了深入研究导致RNA表达波动的潜在调控因素,我们先对参与基因表达转录后调控的miRNA进行了进一步的分析。发现在E14d之前,表达下调的miRNA数量显著增加,占到所有差异表达的miRNA的76%(见附录A中的表S16)。使用miRWalk2.0数据库[30]进行的分析显示,下调的miRNA在RNA聚合酶II启动子转录延伸的负调控(negative regulation of transcription elongation from RNA polymerase II promoter, P < 0.01)、DNA模板转录的负调控(negative regulation of DNA-templated transcription, P < 0.01)和转录调控区DNA结合(transcription regulatory region DNA binding, P < 0.01)等功能中显著富集(见附录A中的表S17)。大量下调的miRNA可能会降低其对mRNA的总体降解水平[图3(a)]。piRNA可以通过Piwi-piRNA相互作用来介导转录沉默[31]。我们发现,显著下调的piRNA的比例从E9d开始直到E14d不断升高[见图3(a)中的橙色线、附录A中的表S17],很可能削弱了Piwi-piRNA复合物沉默基因组的能力。
《图3》
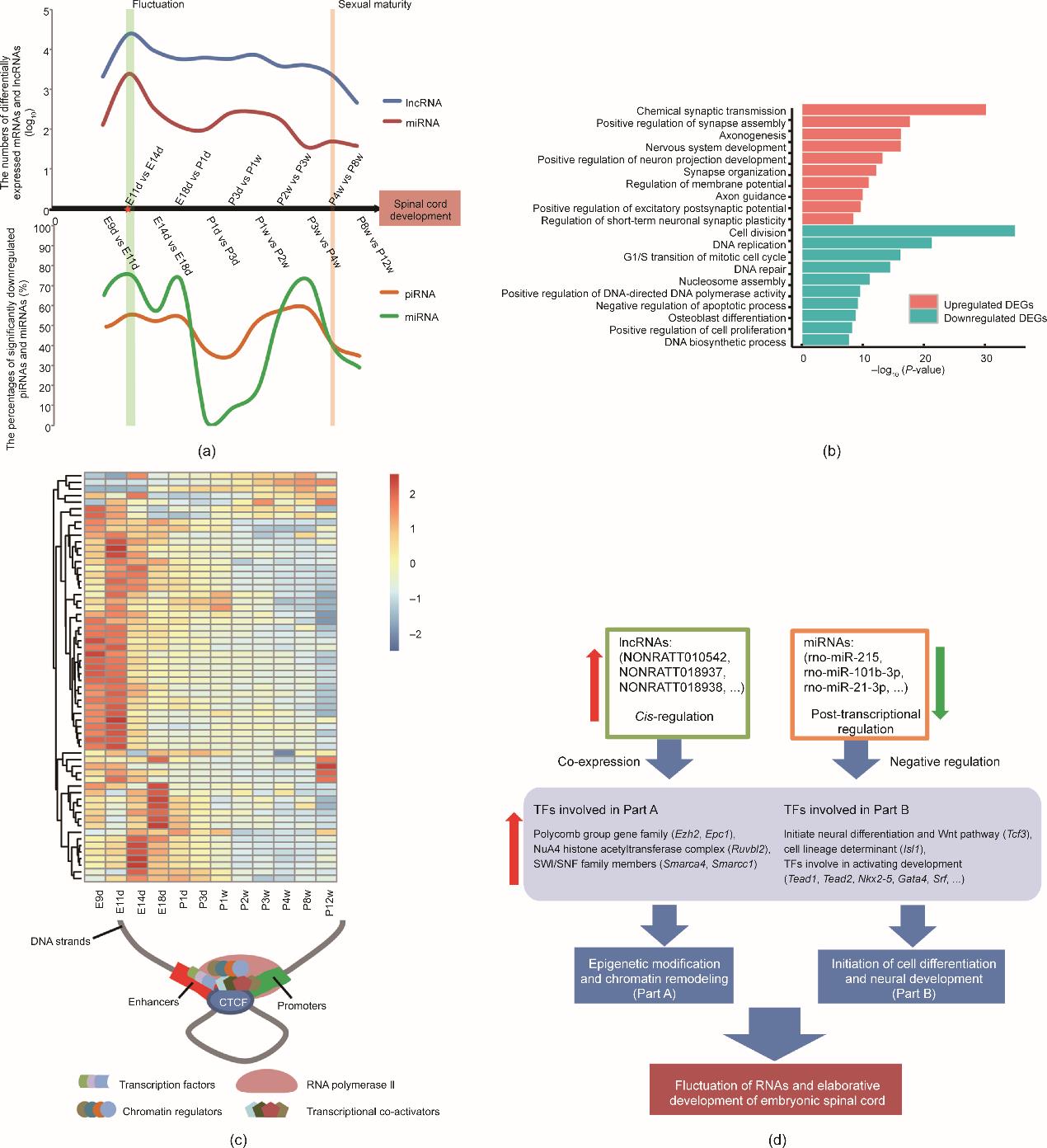
图3 大鼠脊髓发育过程中的RNA波动现象和潜在的调控因子。
除了miRNA和piRNA的改变外,我们还鉴定到了271个作用于该时期DEG上游的TF(见附录A中的表S19),其中57个参与了增强子结合过程(见附录A中的表S20)。深入的表达聚类分析表明,在RNA表达波动之前,这57个转录因子中约有80%已经呈现高水平表达[图3(a)中的热图]。通过毗邻的lncRNA来顺式(cis)调控蛋白编码基因也是一类常见的调控现象[32]。我们从57个参与增强子结合的TF中筛选出13个有上游lncRNA cis调控信息的TF,发现这27个lncRNA(其中50%为已知lncRNA)与其靶向的TF基因呈现出共表达的趋势(见附录A中的图S4、表S21)。有趣的是,靶向这13个TF的miRNA在同时期大部分都处于低表达的状态。此外,这13个TF根据其分子功能可分为两部分:Part A (Ezh2、Epc1、Ruvbl2、Smarca4, Smarcc1),其中主要包括参与表观遗传重编程和染色质重构的转录因子;Part B (Tcf3, Isl1, Tead1, Tead2, Nkx2-5),主要在细胞分化和神经发育的起始过程中发挥作用[图3(d)]。这些潜在的上游调节因子(miRNA、lncRNA和TF)可能协同促进mRNA的波动,而这种波动可能参与脊髓模式和脊髓组织区域化的形成过程。
《3.3. 可变剪接事件的阶段特异性》
3.3. 可变剪接事件的阶段特异性
我们分析了脊髓发育过程中从E9d到P12w的五种主要可变剪接形式[3′端可变剪接(alternative 3′splice site, A3SS)、5′端可变剪接(alternative 5′splice site, A5SS)、外显子互斥(mutually exclusive exon, MXE)、内含子保留(retained intron, RI)、外显子跳跃(skipped exon, SE)]。并观察差异性可变剪接事件(differential alternative splicing event, DASE)的时间模式,大多数是SE事件(占所有事件的51.4%,见附录A中的表S22)[19]。DASE数量分布的两个波峰分别与RNA波动期和新生期相重合[图4(a)]。经历SE剪接事件的基因其生物学功能主要富集在胚胎时期的神经元投射发育过程(neuron projection development, P < 0.01, E11d vs. E14d)和新生期的基于微管的移动过程(microtubule-based movement, P < 0.05, E18d vs. P1d)中。而那些经历RI剪接事件的基因其生物学功能主要富集在胚胎时期的动作电位期间的膜去极化过程(membrane depolarization during action potential, P < 0.01, E11d vs. E14d)和新生期的核质细胞成分(cellular components of nucleoplasm, P < 0.01, E18d vs. P1d)中。其他三种可变剪接事件(MEX、A5SS和A3SS)随着时间的推移表现出相对稳定的模式,但在E11d到E14d时仍有轻微的起伏[图4(a)]。特别地,我们注意到组成郎飞结及其附属结构Paranode和Juxtaparanode的主要功能性大分子组分的基因正好都处在DASE分布的两个主要波峰之中[E11d vs E14d, E18d vs P1d;见图4(b)、附录A中的表S23] [33]。郎飞结及其附属结构是有颌类脊椎动物在进化过程产生的,同时,可变剪接在发育和进化过程中发挥着重要的作用[34‒35]。因此,DASE结果表明,发育中的部分可变剪接可能是促进郎飞结发育成熟的驱动力之一。
《图4》
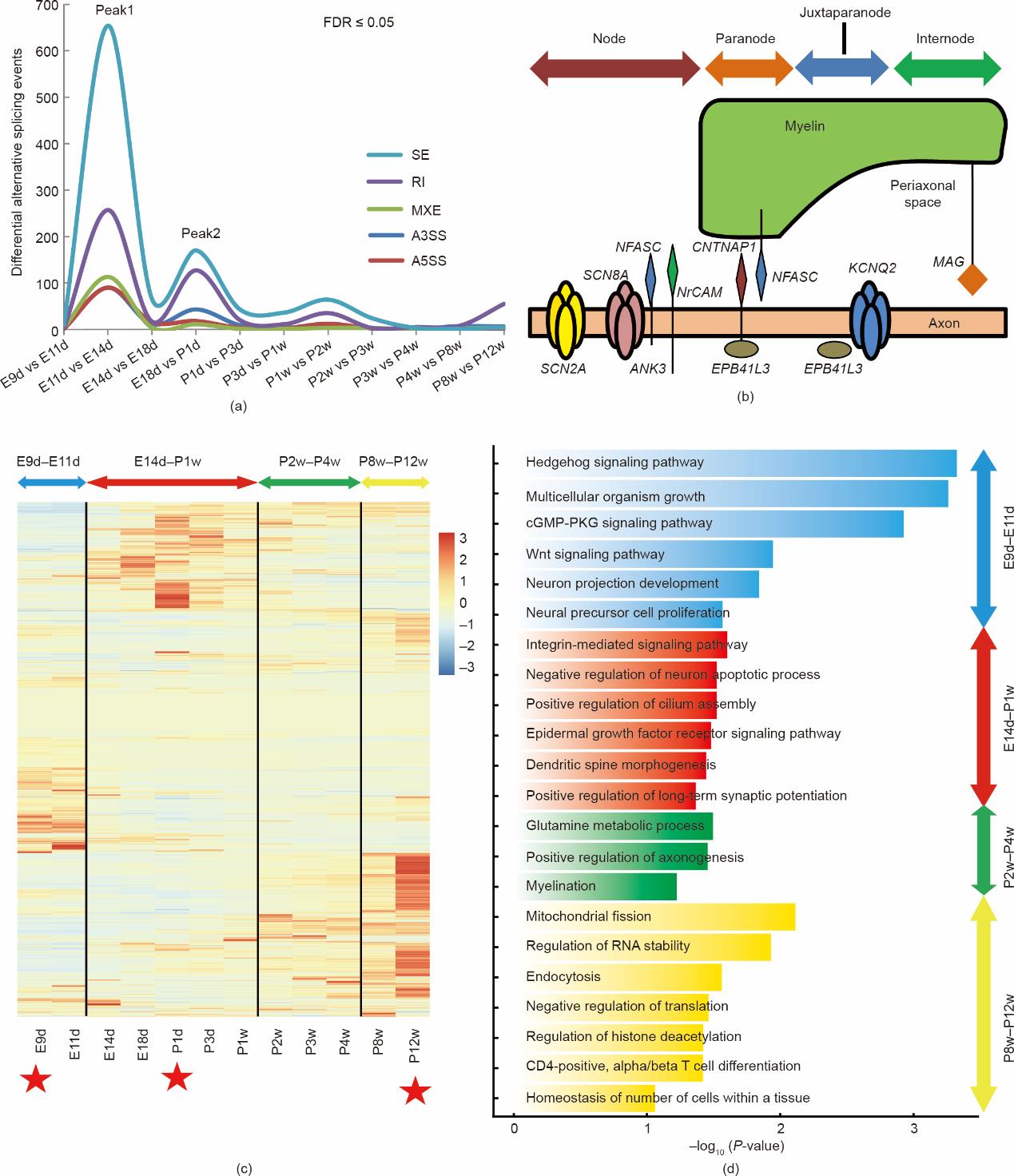
图4 大鼠脊髓发育过程中选择性剪接事件的独特模式。
进一步地,我们将脊髓发育转录组数据与已知可变剪接数据库的比较分析显示,具有全新的可变剪接的基因主要分布在E9d、P1d和P12w附近[图4(c)]。我们以这三个时间点为参考,将大鼠脊髓发育过程分为以下几个阶段[图4(c)]:①脊髓早期发育阶段(E9d~E11d);②胚胎后期和新生期的脊髓发育(E14d~P1w);③幼年期的脊髓发育(P2w~P4w);④成年期(P8w~P12w)。我们还对这些具有全新可变剪接特征基因的模式进行了WGCNA分析,获得了27个共表达模块(见附录A中的图S5)。这27个模块的功能逐渐从胚胎发育、干细胞维持和细胞分化转向与髓鞘形成、免疫系统和稳态维持相关的功能[图4(d)]。这些数据为进一步研究新的剪接变异提供了有价值的资源。此外,与可变剪接密切相关的RNA结合蛋白(RBP)也显示出与全新可变剪接基因类似的时间聚类模式(见附录A中的图S6)。作为蛋白质的RBP也可以通过相分离来响应刺激、温度、pH值或其他微环境信号的变化,并从可溶性蛋白转变为不溶性蛋白,形成无膜细胞器[36]。我们进一步预测到24个具有内在无序区域(IDR, involved in phase separation [37];见附录A中的图S7)的RBP在E9d和E11d高表达(见附录A中的图S8),这些RBP可能参与了对脊髓微环境的响应。CTCF在三维基因组的结构调控中具有多种关键功能,包括选择性剪接和调控功能相关基因群的转录[38‒39],在大鼠脊髓发育过程中CTCF也在E9d和E11d高表达(见附录A中的表S24)。
《3.4. 天生免疫和内在生长能力》
3.4. 天生免疫和内在生长能力
WGCNA的分析发现,模块M7 [图1(d)]在免疫应答相关基因中显著富集(immune response, P < 0.01)。M7中基因的表达在出生后逐渐上调,在性成熟时达到峰值[图1(d)中的热图]。我们还发现许多上调的TF在P3d后与先天免疫相关。由于小胶质细胞是中枢神经系统唯一的原位免疫细胞[40],我们分析了参与小胶质细胞增殖、发育和激活的基因,发现这些基因的表达模式(尤其是小胶质细胞标志物Cx3cr1 [41](见附录A中的表S25)与M7一致(见附录A中的图S9)。
为了更好地理解先天免疫与脊髓内在生长能力的潜在联系。我们仔细调查了在不同的发育阶段可能涉及的因素,并建立了一个早期框架图[见图5(a)、附录A中的表S26]。在胚胎时期(E9d~E18d),脊髓组织转录组中大量活跃的干性维持和细胞去分化基因持续表达(见附录A中的图S1、图S10、表S26),免疫系统发育非常缓慢,赋予了胚胎脊髓天然而强大的内在生长能力。然而,这些有助于内在生长能力的基因在幼年和青春期(P1d~P4w)下调(见附录A中的图S9、图S10)。同时,促进小胶质细胞从M0向M2极化(M2型小胶质细胞可抑制细胞凋亡,促进再生[42])的部分调节因子(包括TF、miRNA和lncRNA,如Kdm6b、miR-124和GAS5等)表现出易化小胶质细胞M2型极化的潜在倾向(见附录A中的表S26)。Toll样受体(TLR)表达水平的低水平增长有助于炎症控制和伤口愈合[43‒45]。数据显示大多数TLR的表达量从P1d到P4w缓慢地增长(见附录A中的图S11),提示了TLR对内在生长能力可能有潜在的正面影响。因此,在这一时期,不利于再生的免疫系统相关呼吸暴发(immune system-relevant respiratory burst, RB)作用可以得到一定程度的抑制,从而维持部分脊髓的内在生长能力。在大鼠性成熟(P8w~P12w)之后,我们发现与脊髓内在生长能力相关的基因表达发生了新的变化(见附录A中的表S26),特别是与先天免疫发育成熟相关的基因表达发生了较大的改变(见附录A中的图S9)。一些促进小胶质细胞从M0型向M1型极化的调节因子(如TLR、miR-21和miR-101)的表达变化可能易化特定条件下小胶质细胞向不利于愈伤的M1型极化(见附录A中的表S26)。白细胞介素4受体(IL4R)及其配体IL4能促进M0型小胶质细胞重编程为M2型小胶质细胞[42]。数据显示,在P12w时Il4r显著下调(见附录A中的表S27),这可能会影响IL4对M0型小胶质细胞的重编程,减少潜在的M2型小胶质细胞的数量。一些模式识别受体,如Toll样受体4(TLR4)和CASP4(CASP4),它们可以促进小胶质细胞从M0到M1型的极化[46],转录组数据显示,它们在大鼠出生后呈现持续的高表达(见附录A中的表S27),这可能导致脊髓中潜在的M1型小胶质细胞增加。成年哺乳动物中枢神经系统损伤后,会出现剧烈的小胶质细胞再殖过程[47],产生大量的M1型小胶质细胞,再通过炎症诱发成年脊髓中严重的RB作用而进一步损伤脊髓、阻碍再生。此外,双亮氨酸拉链激酶(DLK)是轴突修复过程中损伤相关信号逆行转运所必需的[48],它在出生后出现显著的下调(见附录A中的表S29),减弱损伤信号的传导,这可能会降低脊髓总体的内在生长能力。综上所述,脊髓发育的转录组数据表明,脊髓中复杂的时间依赖性相互作用可能促成阻碍其内在生长能力建立的恶性循环的逐步形成,进一步导致慢性、进行性的神经退行性改变,最终导致脊髓再生能力的逐步减退[图5(a)]。
《图5》
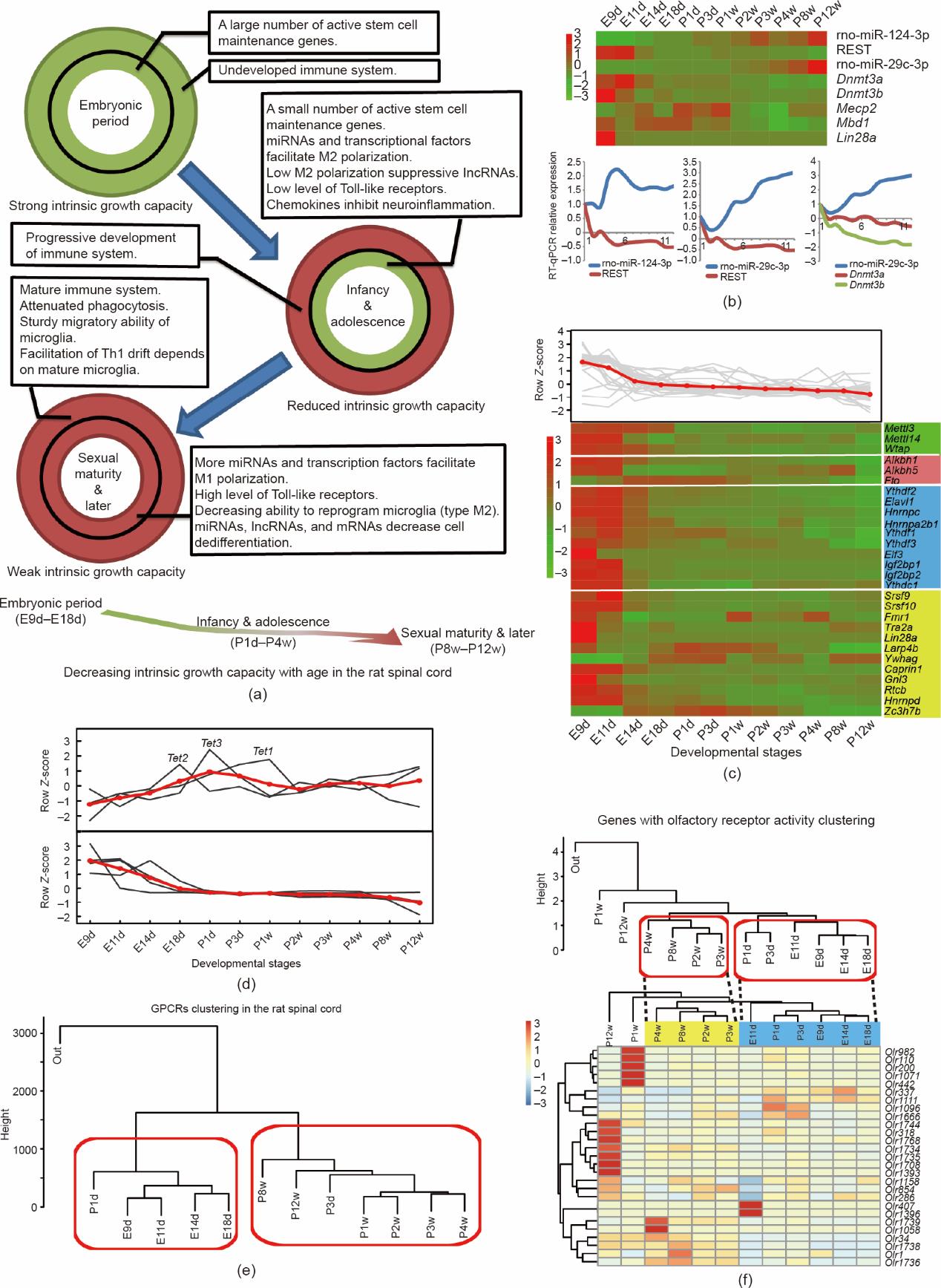
图5 大鼠脊髓发育过程中组织内在生长能力的弱化、表观遗传修饰以及G蛋白偶联受体(GPCR)在发育过程中的主要特征。
《3.5. 表观遗传修饰和G蛋白偶联受体》
3.5. 表观遗传修饰和G蛋白偶联受体
表观遗传修饰是神经元发育的重要调控因素。我们分析了在脊髓发育过程中的三种表观遗传修饰:CpH甲基化(non-CpG,其中,“H”可以是腺苷、胞嘧啶或胸腺嘧啶)、N6-methyladenosine (m6A)甲基化、DNA的主动去甲基化。首先,CpH甲基化是中枢神经系统成熟的关键调控因子,并由DNA甲基转移酶3α(DNMT3A)来负责维持[49‒50]。RE1沉默转录因子(REST)通过miR-29c-3p来调控Dnmt3a [51]。大鼠脊髓发育过程中,Dnmt3a在出生前表现出缓慢而持续的表达模式,这可能导致在脊髓发育早期CpH甲基化的潜在积累[图5(b)] [49]。其次,m6A甲基化与可变剪接和mRNA的稳定性有关[52‒53]。我们的数据显示,发育过程中大多数参与m6A甲基化的基因(包括Writers、Erasers、Readers和相关的RBP)表现出缓慢下调的趋势[图5(c)],这表明m6A甲基化可能在发育过程中处于一种时间依赖性的总体动态平衡状态[54‒56]。再次,10~11易位(TET)蛋白家族的表达表明,活跃的DNA去甲基化事件可能发生在围产期前后[见图5(d)和附录A中的表S30]。5个主要的TET调节因子(Lin28a, Vprbp, Ogt, Parp1, Sin3a)[57‒61]在胚胎周期中下降[图5(d)]。这些数据表明,活性DNA去甲基化可能在围产期的基因调控中发挥关键作用。最后,ten-eleven translocation (TET)蛋白家族的表达表明,脊髓发育过程中的DNA主动去甲基化事件可能发生在围产期前后[见图5(d)和附录A中的表S30]。部分主要的TET调节因子(Lin28a, Vprbp, Ogt, Parp1,Sin3a)[57-61]在胚胎期逐渐下调[图5(d)],这些数据表明,DNA主动去甲基化可能在发育环境改变剧烈的围产期发育调控中发挥潜在的重要作用。
G蛋白偶联受体(GPCR)是哺乳动物中最大的受体超家族[62]。在大鼠脊髓发育过程中,一共注释到883个具有GPCR活性的基因(GO:0004930)。基于12个时间点GPCR表达的分层聚类分析显示出两个主要的时间簇:E9d-P1d簇和P3d-P12w簇[图5(e)]。GPCR也可进一步细分为三个基因簇(见附录A中的图S12),其共同点是嗅觉转导通路(KEGG PATHWAY: rno04740)、神经活性配体-受体相互作用通路(KEGG PATHWAY: rno04080)和钙信号通路(KEGG PATHWAY:rno04020)在这三个基因簇中均被显著富集(P < 0.01)。先前的研究表明,嗅觉受体(OR)在嗅觉上皮和精子中高度表达,而在其他组织中表达较低[63‒64]。我们发现在新生期前后,大鼠脊髓中421个具有OR活性的基因的表达发生了变化[图5(f)的上侧]。其中27个相对高表达的OR(FPKM ≥ 0.3)也表现出相类似的变化[represented by dotted lines in 图5(f), lower panel]。而先前WCCNA的分析表明,在M6和M10模块中也存在大量的OR,其生物学功能分别是参与胚胎发育和轴突生长。提示OR作为一大类重要的特异性化学受体,其表达可能与发育相关化学物质的识别和轴突导向有潜在关联。
《4. 结论》
4. 结论
综上所述,我们通过大鼠脊髓组织时序转录组测序获得了涵盖脊髓发育过程中的12个时间点(包括胚胎期、新生期、幼年期和成年期)的长期转录组数据。这是目前为止对大鼠脊髓组织发育的最长的时间过程分析。通过生物信息学分析,我们探索了在脊髓发育中发生的分子事件,包括差异基因表达、共表达网络、阶段特异性表达模式和可变剪接事件等。E14d是神经管向脊髓发育过渡的关键时期[65],通过相邻时期的差异基因表达分析,发现在E14d之前大量差异表达基因出现表达波动,提示该时期的基因调控活性相对较高。先天免疫与脊髓的内在生长能力可能存在负相关[66],我们发现先天免疫系统相关基因的确随着发育的持续而逐渐上调,尤其是在出生后最为明显。我们的结果还表明,与郎飞结相关的基因发生了更多的DASE,这提示在有颌脊椎动物中,基因亚型多样性的增加可能有助于郎飞结的进化出现和成熟[67‒68]。这项研究为整个脊髓发育过程中的转录变化提供了宝贵的数据资源,并且我们的时序转录组数据可以与其他研究产生的越来越多的空间转录组和单细胞数据相互补充,为新的组织工程策略的发展提供了更丰富的数据基础,有助于对未来的基础理论和临床研究提供更好的针对策略,本研究也为再生医学和脊髓相关组织工程技术的改进提供了一个整体的参考框架。
Availability of data and materials
RNA-Seq and miRNA-Seq data for different development stages of the rat spinal cord have been deposited in the NCBI database with the BioProject accession tag PRJNA505253.











 京公网安备 11010502051620号
京公网安备 11010502051620号




