
2019, Volume 5, Issue 5
Engineering >> 2019, Volume 5, Issue 5 doi: 10.1016/j.eng.2019.01.017
A Comparative Analysis of the Chloroplast Genomes of Four Salvia Medicinal Plants
a College of Pharmacy, Shandong University of Traditional Chinese Medicine (TCM), Jinan 250355, China
b Institute of Chinese Materia Medica, China Academy of Chinese Medical Sciences, Beijing 100700, China
c State Key Laboratory of Innovative Natural Medicine and TCM Injections, Jiangxi Qingfeng Pharmaceutical Co. Ltd., Ganzhou 341000, China
d Beijing Children's Hospital, Capital Medical University, National Center for Children's Health, Beijing 100045, China
e School of Biological Science, The University of Queensland, Brisbane, QLD 4072, Australia
f College of Pharmacy, Dali University, Dali 671000, China
g Institute of Medicinal Plants, Yunnan Academy of Agricultural Sciences, Kunming 650205, China
Next Previous
Abstract
Herbgenomics is an emerging field of traditional Chinese medicine (TCM) research and development. By combining TCM research with genomics, herbgenomics can help to establish the scientific validity of TCM and bring it into wider usage within the field of medicine. Salvia Linn. is a large genus of Labiatae that includes important medicinal plants. In this herbgenomics study, the complete chloroplast (cp) genomes of two Salvia (S.) spp.—namely, S. przewalskii and S. bulleyana, which are used as a surrogate for S. miltiorrhiza—were sequenced and compared with those of two other reported Salvia spp.—namely, S. miltiorrhiza and S. japonica. The genome organization, gene number, type, and repeat sequences were compared. The annotation results showed that both Salvia plants contain 114 unique genes, including 80 protein-coding, 30 transfer RNA (tRNA), and four ribosomal RNA (rRNA) genes. Repeat sequence analysis revealed 21 direct and 22 palindromic
sequences in both Salvia cp genomes, and 17 and 21 tandem repeats in S. przewalskii and S. bulleyana, respectively. A synteny comparison of the Salvia spp. cp genomes showed a high degree of sequence similarity in the coding regions and a relatively high divergence of the intergenic spacers. Pairwise alignment and singlenucleotide polymorphism (SNP) analyses found some candidate fragments to identify Salvia spp., such as the intergenic region of the trnV-ndhC, trnQ-rps16, atpI-atpH, psbA-ycf3, ycf1, rpoC2, ndhF, matK, rpoB, rpoA and accD genes. All of the results—including the repeat sequences and SNP sites, the inverted repeat (IR) region border, and the phylogenetic analysis—showed that S. przewalskii and S. bulleyana are extremely similar from a genetic standpoint. The cp genome sequences of the two Salvia spp. reported here will pave the way for breeding, species identification, phylogenetic evolution, and cp genetic engineering studies of Salvia medicinal plants.
Keywords
Figures

Fig. 1
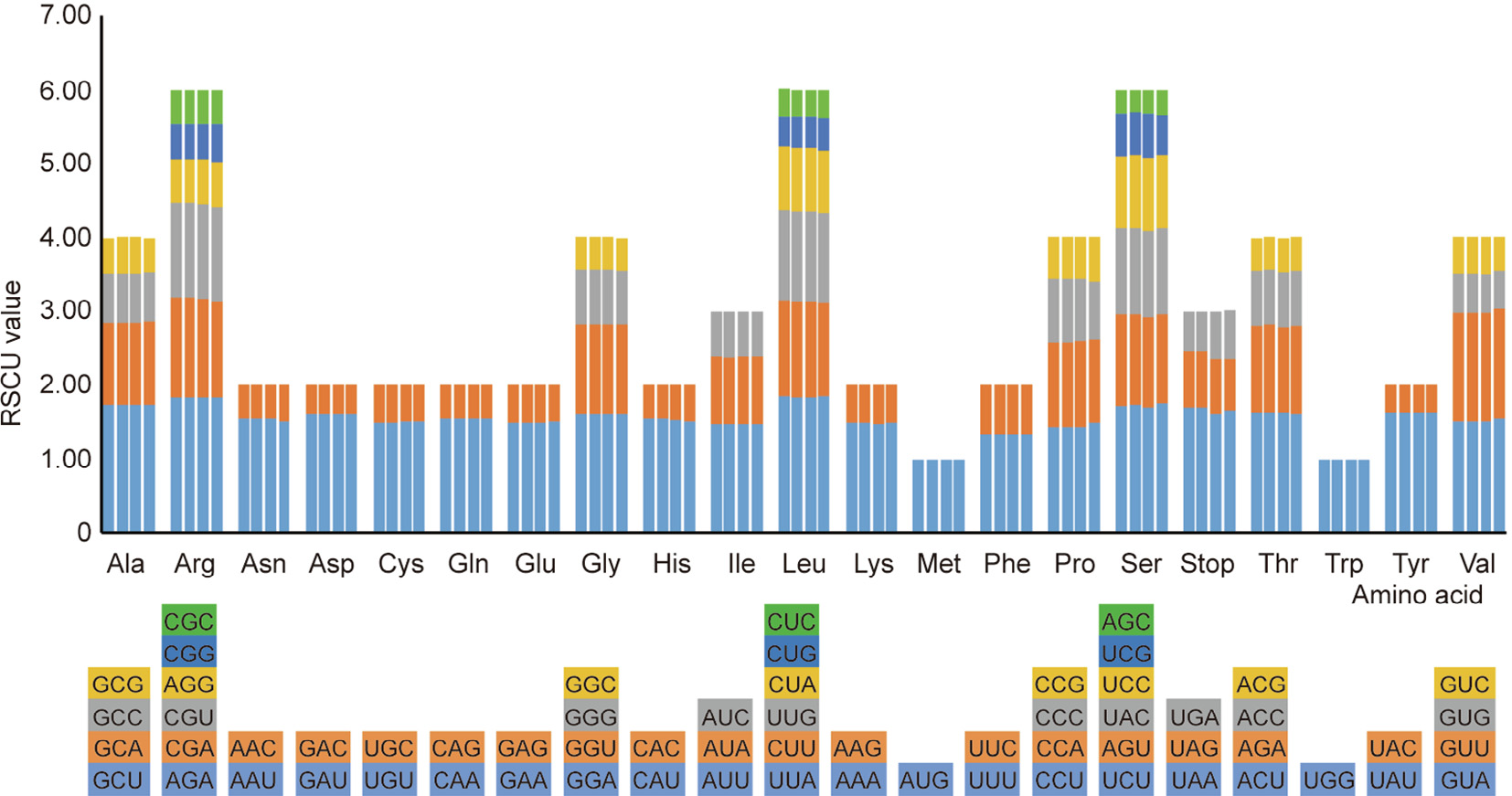
Fig. 2

Fig. 3

Fig. 4
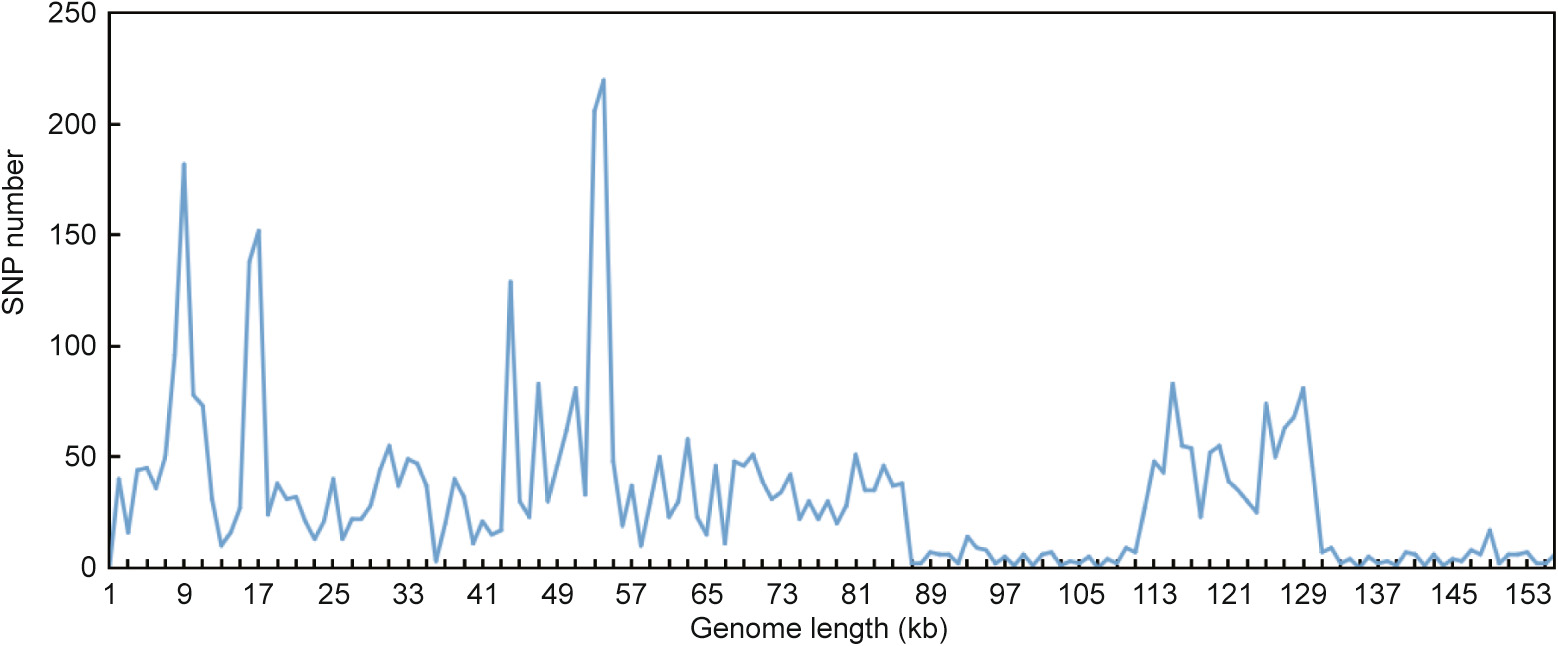
Fig. 5

Fig. 6
References
[ 1 ] Xiao XH, Fang QM, Xia WJ, Yin GP, Su ZW, Qiao CZ. Numerical taxonomy of medicinal Salvia L. and the genuineness of Danshen. J Plant Resour Environ 1997;6(2):17–21. Chinese. link1
[ 2 ] Wang Y, Li D, Zhang Y. Analysis of ITS sequences of some medicinal plants and their related species in Salvia. Yao Xue Xue Bao 2007;42(12):1309–13. Chinese. link1
[ 3 ] Li ZM, Xu SW, Liu PQ. Salvia miltiorrhiza Burge (Danshen): a golden herbal medicine in cardiovascular therapeutics. Acta Pharmacol Sin 2018;39 (5):802–24. link1
[ 4 ] Wang L, Ma R, Liu C, Liu H, Zhu R, Guo S, et al. Salvia miltiorrhiza: a potential red light to the development of cardiovascular diseases. Curr Pharm Des 2017;23 (7):1077–97. link1
[ 5 ] Chen W, Chen GX. Danshen (Salvia miltiorrhiza Bunge): a prospective healing sage for cardiovascular diseases. Curr Pharm Des 2017;23(34):5125–35. link1
[ 6 ] Chinese Pharmacopoeia Commission. Pharmacopoeia of the People’s Republic of China. 2015 ed. Beijing: People’s Medical Publishing; 2015. Appendix 76. link1
[ 7 ] Ren H, Hu X, Liu Y, Dai D, Liu X, Wang Z, et al. Salvia przewalskii extract of total phenolic acids inhibit TLR4 signaling activation in podocyte injury induced by puromycin aminonucleoside in vitro. Ren Fail 2018;40(1):273–9. link1
[ 8 ] Yang Y, Wang ZP, Gao SH, Ren HQ, Zhong RQ, Chen WS. The effects of Salvia przewalskii total phenolic acid extract on immune complex glomerulonephritis. Pharm Biol 2017;55(1):2153–60. link1
[ 9 ] Wang HQ, Yang LX, Chen XY, Yang PF, Chen RY. Chemical constituents from Salvia przewalskii root. J Chin Med Mater 2015;38(6):1197–201. Chinese. link1
[10] Liu X, Liu Y, Yang Y, Xu J, Dai D, Yan C, et al. Antioxidative stress effects of Salvia przewalskii extract in experimentally injured podocytes. Nephron 2016;134 (4):253–71. link1
[11] Li SF, Li JQ, Li XJ, Wang XY, Du PJ, Zhou KP, et al. Pharmacognostical studies on Salvia buleyana Diela. Chin J Ethnomed Ethnopharm 2008;7:18–22. Chinese. link1
[12] Brunkard JO, Runkel AM, Zambryski PC. Chloroplasts extend stromules independently and in response to internal redox signals. Proc Natl Acad Sci USA 2015;112(32):10044–9. link1
[13] Poczai P, Hyvönen J. The complete chloroplast genome sequence of the CAM epiphyte Spanish moss (Tillandsia usneoides, Bromeliaceae) and its comparative analysis. PLoS ONE 2017;12(11):e0187199. link1
[14] Douglas SE. Plastid evolution: origins, diversity, trends. Curr Opin Genet Dev 1998;8(6):655–61. link1
[15] Lin CP, Wu CS, Huang YY, Chaw SM. The complete chloroplast genome of Ginkgo biloba reveals the mechanism of inverted repeat contraction. Genome Biol Evol 2012;4(3):374–81. link1
[16] Mo JS, Kim K, Lee MH, Lee JH, Yoon UH, Kim TH. The complete chloroplast genome sequence of Perilla citriodora (Makino) Nakai. Mitochondrial DNA Part A 2017;28(1):131–2. link1
[17] Wicke S, Schneeweiss GM, DePamphilis CW, Müller KF, Quandt D. The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol Biol 2011;76(3–5):273–97. link1
[18] Wang MX, Cui LC, Feng KW, Deng PC, Du XH, Wan FH, et al. Comparative analysis of Asteraceae chloroplast genomes: structural organization, RNA editing and evolution. Plant Mol Biol Report 2015;33(5):1526–38. link1
[19] Raubeson LA, Peery R, Chumley TW, Dziubek C, Fourcade HM, Boore JL, et al. Comparative chloroplast genomics: analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genomics 2007;8(1):174. link1
[20] Wang RJ, Cheng CL, Chang CC, Wu CL, Su TM, Chaw SM. Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol Biol 2008;8(1):36. link1
[21] Zhu A, Guo W, Gupta S, Fan W, Mower JP. Evolutionary dynamics of the plastid inverted repeat: the effects of expansion, contraction, and loss on substitution rates. New Phytol 2016;209(4):1747–56. link1
[22] Li X, Yang Y, Henry RJ, Rossetto M, Wang Y, Chen S. Plant DNA barcoding: from gene to genome. Biol Rev Camb Philos Soc 2015;90(1):157–66. link1
[23] Daniell H, Lin CS, Yu M, Chang WJ. Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol 2016;17(1):134. link1
[24] Cheng L, Li HP, Qu B, Huang T, Tu JX, Fu TD, et al. Chloroplast transformation of rapeseed (Brassica napus) by particle bombardment of cotyledons. Plant Cell Rep 2010;29(4):371–81. link1
[25] Svab Z, Hajdukiewicz P, Maliga P. Stable transformation of plastids in higher plants. Proc Natl Acad Sci USA 1990;87(21):8526–30. link1
[26] Rˇepková J. Potential of chloroplast genome in plant breeding. Czech J Genet Plant Breed 2010;46(3):103–13. link1
[27] Chen S, Xu J, Liu C, Zhu Y, Nelson DR, Zhou S, et al. Genome sequence of the model medicinal mushroom Ganoderma lucidum. Nat Commun 2012;3(1):913. link1
[28] Huang ZH, Xu J, Xiao SM, Liao BS, Gao Y, Zhai CC, et al. Comparative optical genome analysis of two pangolin species: Manis pentadactyla and Manis javanica. GigaScience 2016;5(1):1–5. link1
[29] Xu J, Chu Y, Liao B, Xiao S, Yin Q, Bai R, et al. Panax ginseng genome examination for ginsenoside biosynthesis. GigaScience 2017;6(11):1–15. link1
[30] Qian J, Song J, Gao H, Zhu Y, Xu J, Pang X, et al. The complete chloroplast genome sequence of the medicinal plant Salvia miltiorrhiza. PLoS ONE 2013;8 (2):e57607. link1
[31] Xu H, Song J, Luo H, Zhang Y, Li Q, Zhu Y, et al. Analysis of the genome sequence of the medicinal plant Salvia miltiorrhiza. Mol Plant 2016;9(6):949–52. link1
[32] He Y, Xiao H, Deng C, Xiong L, Yang J, Peng C. The complete chloroplast genome sequences of the medicinal plant Pogostemon cablin. Int J Mol Sci 2016;17 (6):820. link1
[33] Li XW, Gao HH, Wang YT, Song JY, Henry R, Wu HZ, et al. Complete chloroplast genome sequence of Magnolia grandiflora and comparative analysis with related species. Sci China Life Sci 2013;56(2):189–98. link1
[34] Shen X, Wu M, Liao B, Liu Z, Bai R, Xiao S, et al. Complete chloroplast genome sequence and phylogenetic analysis of the medicinal plant Artemisia annua. Molecules 2017;22(8):1330. link1
[35] Jiang D, Zhao Z, Zhang T, Zhong W, Liu C, Yuan Q, et al. The chloroplast genome sequence of Scutellaria baicalensis provides insight into intraspecific and interspecific chloroplast genome diversity in Scutellaria. Genes 2017;8(9):227. link1
[36] Vining KJ, Johnson SR, Ahkami A, Lange I, Parrish AN, Trapp SC, et al. Draft genome sequence of Mentha longifolia and development of resources for mint cultivar improvement. Mol Plant 2017;10(2):323–39. link1
[37] Guo H, Liu J, Luo L, Wei X, Zhang J, Qi Y, et al. Complete chloroplast genome sequences of Schisandra chinensis: genome structure, comparative analysis, and phylogenetic relationship of basal angiosperms. Sci China Life Sci 2017;60 (11):1286–90. link1
[38] Mariotti R, Cultrera NG, Díez CM, Baldoni L, Rubini A. Identification of new polymorphic regions and differentiation of cultivated olives (Olea europaea L.) through plastome sequence comparison. BMC Plant Biol 2010;10(1):211. link1
[39] Yi DK, Kim KJ. Complete chloroplast genome sequences of important oilseed crop Sesamum indicum L. PLoS ONE 2012;7(5):e35872. link1
[40] Lu C, Shen Q, Yang J, Wang B, Song C. The complete chloroplast genome sequence of Safflower (Carthamus tinctorius L). Mitochondrial DNA, Part A 2016;27(5):3351–3. link1
[41] Wu JY, Xiao JF, Wang LP, Zhong J, Yin HY, Wu SX, et al. Systematic analysis of intron size and abundance parameters in diverse lineages. Sci China Life Sci 2013;56(10):968–74. link1
[42] Xu J, Feng D, Song G, Wei X, Chen L, Wu X, et al. The first intron of rice EPSP synthase enhances expression of foreign gene. Sci China Life Sci 2003;46 (6):561. link1
[43] Yan J, Zhang Q, Yin P. RNA editing machinery in plant organelles. Sci China Life Sci 2018;61(2):162–9. link1
[44] Chen C, Bundschuh R. Systematic investigation of insertional and deletional RNA-DNA differences in the human transcriptome. BMC Genomics 2012;13 (1):616. link1
[45] Knoop V. When you can’t trust the DNA: RNA editing changes transcript sequences. Cell Mol Life Sci 2011;68(4):567–86. link1
[46] Grennan AK. To thy proteins be true: RNA editing in plants. Plant Physiol 2011;156(2):453–4. link1
[47] Lutz KA, Maliga P. Chapter 23—transformation of the plastid genome to study RNA editing. Methods Enzymol 2007;424:501–18. link1
[48] Asaf S, Khan AL, Khan MA, Waqas M, Kang SM, Yun BW, et al. Chloroplast genomes of Arabidopsis halleri ssp. gemmifera and Arabidopsis lyrata ssp. petraea: structures and comparative analysis. Sci Rep 2017;7(1):7556. link1
[49] Yin D, Wang Y, Zhang X, Ma X, He X, Zhang J. Development of chloroplast genome resources for peanut (Arachis hypogaea L.) and other species of Arachis. Sci Rep 2017;7(1):11649. link1
[50] Zhou T, Wang J, Jia Y, Li W, Xu F, Wang X. Comparative chloroplast genome analyses of species in Gentiana section Cruciata (Gentianaceae) and the development of authentication markers. Int J Mol Sci 2018;19(7):1962. link1
[51] Flannery ML, Mitchell FJ, Coyne S, Kavanagh TA, Burke JI, Salamin N, et al. Plastid genome characterisation in Brassica and Brassicaceae using a new set of nine SSRs. Theor Appl Genet 2006;113(7):1221–31. link1
[52] Shi QH, Yao ZP, Zhang H, Xu L, Dai PH. Comparison of four methods of DNA extraction from chickpea. J Xinjiang Agric Univ 2009;1:64–7. Chinese. link1
[53] Kaila T, Chaduvla PK, Rawal HC, Saxena S, Tyagi A, Mithra S, et al. Chloroplast genome sequence of clusterbean (Cyamopsis tetragonoloba L.): genome structure and comparative analysis. Genes 2017;8(9):212. link1
[54] Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014;30(15):2114–20. link1
[55] Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol 1990;215(3):403–10. link1
[56] Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience 2015;4 (1):30. Corrected and republished from GigaScience 2012;1(1):18. link1
[57] Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. Scaffolding preassembled contigs using SSPACE. Bioinformatics 2011;27(4):578–9. link1
[58] Liu C, Shi L, Zhu Y, Chen H, Zhang J, Lin X, et al. CpGAVAS, an integrated web server for the annotation, visualization, analysis and GenBank submission of completely sequenced chloroplast genome sequences. BMC Genomics 2012;13 (1):715. link1
[59] Misra S, Harris N. Using Apollo to browse and edit genome annotations. Curr Protoc Bioinformatics 2006;12(1);9.5.1–28. link1
[60] Schattner P, Brooks AN, Lowe TM. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res 2005;33 (Suppl 2):W686–9. link1
[61] Lohse M, Drechsel O, Bock R. Organellar genome DRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr Genet 2007;52(5–6):267–74. link1
[62] Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 2013;30(12):2725–9. link1
[63] Jansen RK, Kaittanis C, Saski C, Lee SB, Tomkins J, Alverson AJ, et al. Phylogenetic analyses of Vitis (Vitaceae) based on complete chloroplast genome sequences: effects of taxon sampling and phylogenetic methods on resolving relationships among rosids. BMC Evol Biol 2006;6(1):32. link1
[64] Kurtz S, Choudhuri JV, Ohlebusch E, Schleiermacher C, Stoye J, Giegerich R. REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res 2001;29(22):4633–42. link1
[65] Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 1999;27(2):573–80. link1
[66] Thiel T, Michalek W, Varshney RK, Graner A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor Appl Genet 2003;106(3):411–22. link1
[67] Frazer KA, Pachter L, Poliakov A, Rubin EM, Dubchak I. VISTA: computational tools for comparative genomics. Nucleic Acids Res 2004;32(Suppl 2):W273–9. link1
[68] Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 2011;28(10):2731–9. link1
[69] Hollingsworth PM, Forrest LL, Spouge JL, Hajibabaei M, Ratnasingham S, van der Bank M, et al. A DNA barcode for land plants. Proc Natl Acad Sci USA 2009;106(31):12794–7. link1
[70] Yang P, Shen WH, Shi JM, Chen XY, Zhang KK, Guan P. Identification of DNA barcoding of common medicinal plants in Lamiaceae. Chin Tradit Herb Drugs 2017;7:1397–402. Chinese. link1
[71] Gao L, Wang B, Wang ZW, Zhou Y, Su YJ, Wang T. Plastome sequences of Lygodium japonicum and Marsilea crenata reveal the genome organization transformation from basal ferns to core leptosporangiates. Genome Biol Evol 2013;5(7):1403–7. link1
[72] Fleischmann TT, Scharff LB, Alkatib S, Hasdorf S, Schöttler MA, Bock R. Nonessential plastid-encoded ribosomal proteins in tobacco: a developmental role for plastid translation and implications for reductive genome evolution. Plant Cell 2011;23(9):3137–55. link1
[73] Ueda M, Nishikawa T, Fujimoto M, Takanashi H, Arimura S, Tsutsumi N, et al. Substitution of the gene for chloroplast RPS16 was assisted by generation of a dual targeting signal. Mol Biol Evol 2008;25(8):1566–75. link1
[74] Hebert PD, Cywinska A, Ball SL, DeWaard JR. Biological identifications through DNA barcodes. Proc R Soc B 2003;270(1512):313–21. link1
[75] Song Y, Chen Y, Lv J, Xu J, Zhu S, Li MF, et al. Development of chloroplast genomic resources for species discrimination. Front Plant Sci 2017;8:1854. link1
[76] Dong W, Xu C, Li C, Sun J, Zuo Y, Shi S, et al. ycf1, the most promising plastid DNA barcode of land plants. Sci Rep 2015;5(1):8348. link1
[77] Zhang L, Yang Z, Huang X, Li J, Wan D. Study on the leaf epidermal structural characters of Salvia miltiorrhiza and Salvia from Sichuan. J Sichuan Univ Nat Sci 2008;45(3):674–80. Chinese. link1
[78] Wang T, Liu SY, Wang L, Wang HY, Zhang L. Anatomical characteristics of laminae and petioles of 11 species of Salvia and their taxonomic significance. China J Chin Mater Med 2014;39(14):2629–34. Chinese. link1
[79] Wang Y, Jiang K, Wang L, Han D, Yin G, Wang J, et al. Identification of Salvia species using high-performance liquid chromatography combined with chemical pattern recognition analysis. J Sep Sci 2018;41(3):609–17. link1
[80] Skała E, Wysokin´ sk H. Tanshinone production in roots of micropropagated Salvia przewalskii maxim. Z Naturforsch C 2005;60(7–8):583–6. link1











 京公网安备 11010502051620号
京公网安备 11010502051620号




