
2015, Volume 1, Issue 2
Engineering >> 2015, Volume 1, Issue 2 doi: 10.15302/J-ENG-2015062
Bioprinting-Based High-Throughput Fabrication of Three-Dimensional MCF-7 Human Breast Cancer Cellular Spheroids
1 State Key Laboratory for Strength and Vibration of Mechanical Structures, School of Aerospace, Xi'an Jiaotong University, Xi'an 710049, China
2 Bioinspired Engineering and Biomechanics Center (BEBC), Xi'an Jiaotong University, Xi'an 710049, China
3 The Key Laboratory of Biomedical Information Engineering of the Ministry of Education, School of Life Science and Technology, Xi'an Jiaotong University, Xi'an 710049, China
Next Previous
Abstract
Cellular spheroids serving as three-dimensional (3D) in vitro tissue models have attracted increasing interest for pathological study and drug-screening applications. Various methods, including microwells in particular, have been developed for engineering cellular spheroids. However, these methods usually suffer from either destructive molding operations or cell loss and non-uniform cell distribution among the wells due to two-step molding and cell seeding. We have developed a facile method that utilizes cell-embedded hydrogel arrays as templates for concave well fabrication and in situ MCF-7 cellular spheroid formation on a chip. A custom-built bioprinting system was applied for the fabrication of sacrificial gelatin arrays and sequentially concave wells in a high-throughput, flexible, and controlled manner. The ability to achieve in situ cell seeding for cellular spheroid construction was demonstrated with the advantage of uniform cell seeding and the potential for programmed fabrication of tissue models on chips. The developed method holds great potential for applications in tissue engineering, regenerative medicine, and drug screening.
Keywords
MCF-7 cellular spheroids ; bioprinting ; hydrogels ; concave wells ; tissue on a chip
Figures
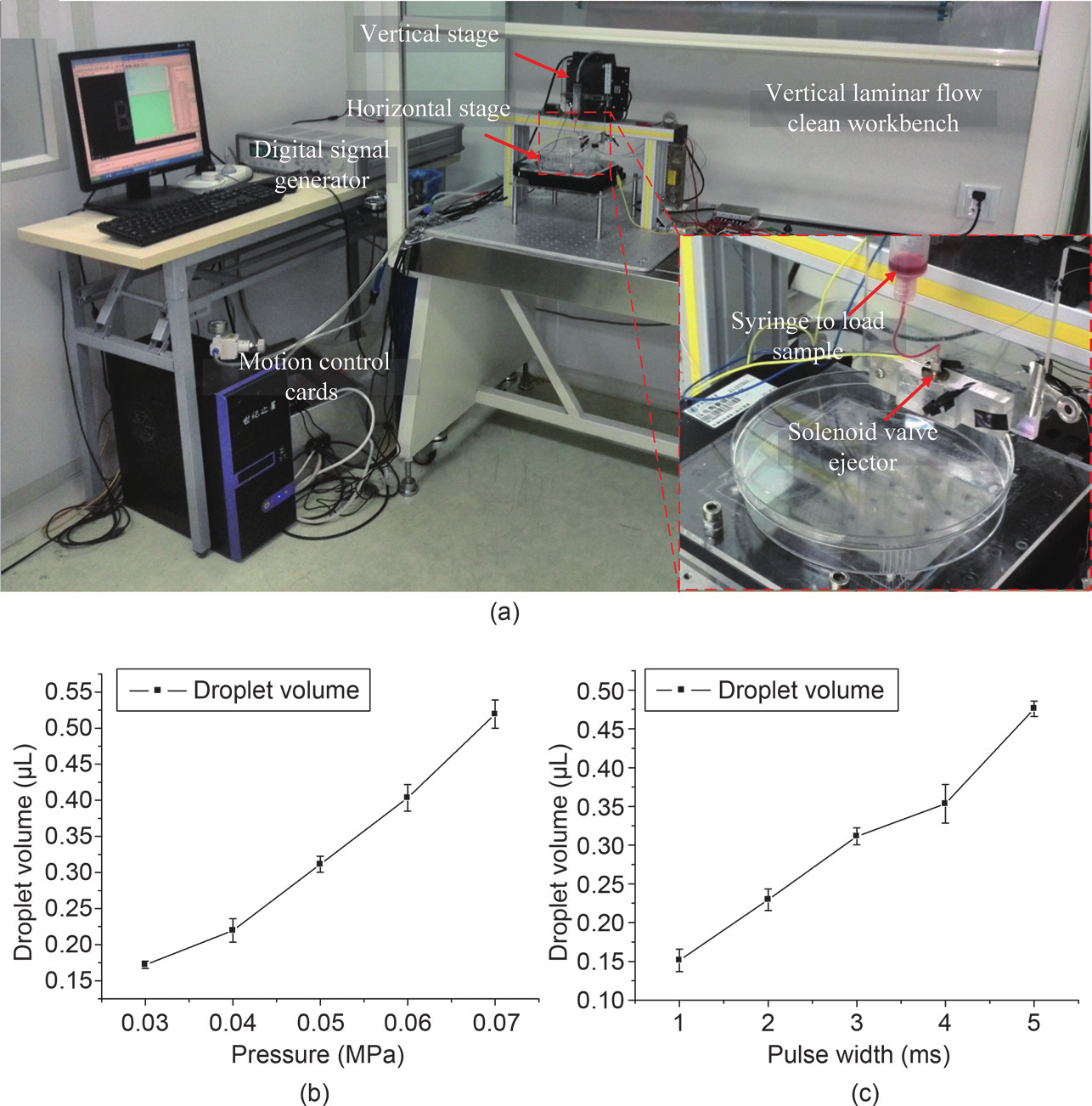
Fig. 1

Fig. 2

Fig. 3
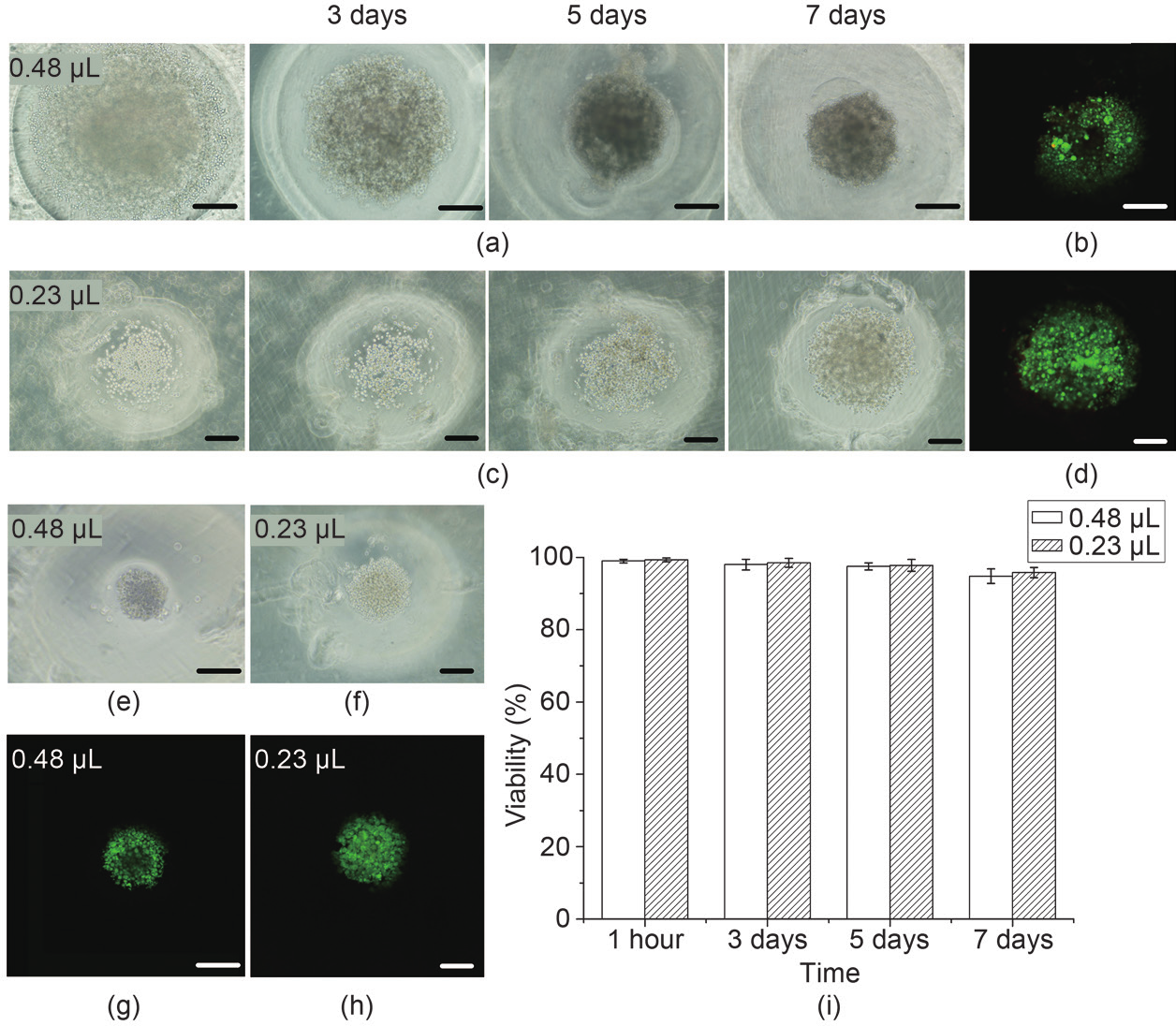
Fig. 4
References
[ 1 ] T. M. Achilli, J. Meyer, J. R. Morgan. Advances in the formation, use and understanding of multi-cellular spheroids. Expert Opin. Biol. Ther., 2012, 12(10): 1347−1360 link1
[ 2 ] M. Rimann, U. Graf-Hausner. Synthetic 3D multicellular systems for drug development. Curr. Opin. Biotechnol., 2012, 23(5): 803−809 link1
[ 3 ] L. Wang, Engineering three-dimensional cardiac microtissues for potential drug screening applications. Curr. Med. Chem., 2014, 21(22): 2497−2509 link1
[ 4 ] J. Rouwkema, J. de Boer, C. A. van Blitterswijk. Endothelial cells assemble into a 3-dimensional prevascular network in a bone tissue engineering construct. Tissue Eng., 2006, 12(9): 2685−2693 link1
[ 5 ] E. Fennema, N. Rivron, J. Rouwkema, C. van Blitterswijk, J. de Boer. Spheroid culture as a tool for creating 3D complex tissues. Trends Biotechnol., 2013, 31(2): 108−115 link1
[ 6 ] K. Takayama, 3D spheroid culture of hESC/hiPSC-derived hepatocyte-like cells for drug toxicity testing. Biomaterials, 2013, 34(7): 1781−1789 link1
[ 7 ] P. R. Baraniak, T. C. McDevitt. Scaffold-free culture of mesenchymal stem cell spheroids in suspension preserves multilineage potential. Cell Tissue Res., 2012, 347(3): 701−711 link1
[ 8 ] A. P. Napolitano, Scaffold-free three-dimensional cell culture utilizing micromolded nonadhesive hydrogels. Biotechniques, 2007, 43(4): 494, 496−500
[ 9 ] D. M. Dean, J. R. Morgan. Cytoskeletal-mediated tension modulates the directed self-assembly of microtissues. Tissue Eng. Part A, 2008, 14(12): 1989−1997 link1
[10] J. Liu, Soft fibrin gels promote selection and growth of tumorigenic cells. Nat. Mater., 2012, 11(8): 734−741 link1
[11] J. Friedrich, C. Seidel, R. Ebner, L. A. Kunz-Schughart. Spheroid-based drug screen: Considerations and practical approach. Nat. Protoc., 2009, 4(3): 309−324 link1
[12] H. F. Chan, Y. Zhang, Y. P. Ho, Y. L. Chiu, Y. Jung, K. W. Leong. Rapid formation of multicellular spheroids in double-emulsion droplets with controllable microenvironment. Sci. Rep., 2013, 3: 3462
[13] F. Langenbach, Generation and differentiation of microtissues from multipotent precursor cells for use in tissue engineering. Nat. Protoc., 2011, 6(11): 1726−1735 link1
[14] M. Inamori, H. Mizumoto, T. Kajiwara. An approach for formation of vascularized liver tissue by endothelial cell-covered hepatocyte spheroid integration. Tissue Eng. Part A, 2009, 15(8): 2029−2037 link1
[15] S. F. Wong, D. Y. No, Y. Y. Choi, D. S. Kim, B. G. Chung, S. H. Lee. Concave microwell based size-controllable hepatosphere as a three-dimensional liver tissue model. Biomaterials, 2011, 32(32): 8087−8096 link1
[16] D. Huh, B. D. Matthews, A. Mammoto, M. Montoya-Zavala, H. Y. Hsin, D. E. Ingber. Reconstituting organ-level lung functions on a chip. Science, 2010, 328(5986): 1662−1668 link1
[17] D. Huh, Y. S. Torisawa, G. A. Hamilton, H. J. Kim, D. E. Ingber. Microengineered physiological biomimicry: Organs-on-chips. Lab Chip, 2012, 12(12): 2156−2164 link1
[18] G. Wang, Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat. Med., 2014, 20(6): 616−623 link1
[19] R. A. Rezende, Scalable biofabrication of tissue spheroids for organ printing. Procedia CIRP, 2013, 5: 276−281 link1
[20] R. J. Thomas, The effect of three-dimensional co-culture of hepatocytes and hepatic stellate cells on key hepatocyte functions in vitro. Cells Tissues Organs (Print), 2005, 181(2): 67−79 link1
[21] Y. C. Tung, A. Y. Hsiao, S. G. Allen, Y. S. Torisawa, M. Ho, S. Takayama. High-throughput 3D spheroid culture and drug testing using a 384 hanging drop array. Analyst, 2011, 136(3): 473−478 link1
[22] G. R. Souza, Three-dimensional tissue culture based on magnetic cell levitation. Nat. Nanotechnol., 2010, 5(4): 291−296 link1
[23] T. Liu, M. Winter, B. Thierry. Quasi-spherical microwells on superhydrophobic substrates for long term culture of multicellular spheroids and high throughput assays. Biomaterials, 2014, 35(23): 6060−6068 link1
[24] S. E. Yeon, Application of concave microwells to pancreatic tumor spheroids enabling anticancer drug evaluation in a clinically relevant drug resistance model. PLoS ONE, 2013, 8(9): e73345 link1
[25] L. Kang, M. J. Hancock, M. D. Brigham, A. Khademhosseini. Cell confinement in patterned nanoliter droplets in a microwell array by wiping. J. Biomed. Mater. Res. A, 2010, 93(2): 547−557
[26] H. Tekin, M. Anaya, M. D. Brigham, C. Nauman, R. Langer, A. Khademhosseini. Stimuli-responsive microwells for formation and retrieval of cell aggregates. Lab Chip, 2010, 10(18): 2411−2418 link1
[27] C. Kim, J. H. Bang, Y. E. Kim, S. H. Lee, J. Y. Kang. On-chip anticancer drug test of regular tumor spheroids formed in microwells by a distributive microchannel network. Lab Chip, 2012, 12(20): 4135−4142 link1
[28] C. Kim, 3-Dimensional cell culture for on-chip differentiation of stem cells in embryoid body. Lab Chip, 2011, 11(5): 874−882 link1
[29] H. C. Moeller, M. K. Mian, S. Shrivastava, B. G. Chung, A. Khademhosseini. A microwell array system for stem cell culture. Biomaterials, 2008, 29(6): 752−763 link1
[30] Y. S. Hwang, B. G. Chung, D. Ortmann, N. Hattori, H. C. Moeller, A. Khademhosseini. Microwell-mediated control of embryoid body size regulates embryonic stem cell fate via differential expression of WNT5a and WNT11. Proc. Natl. Acad. Sci. U.S.A., 2009, 106(40): 16978−16983 link1
[31] Y. Xia, G. M. Whitesides. Soft lithography. Annu. Rev. Mater. Sci., 1998, 28(1): 153−184 link1
[32] Y. Y. Choi, B. G. Chung, D. H. Lee, A. Khademhosseini, J. H. Kim, S. H. Lee. Controlled-size embryoid body formation in concave microwell arrays. Biomaterials, 2010, 31(15): 4296−4303 link1
[33] A. Y. Hsiao, Microfluidic system for formation of PC-3 prostate cancer co-culture spheroids. Biomaterials, 2009, 30(16): 3020−3027 link1











 京公网安备 11010502051620号
京公网安备 11010502051620号




