
《1.高压制造的新型绿色途径》
1.高压制造的新型绿色途径
压力是一种基本的热力学变量,它有许多理想的特性,能控制并增强化学物质和固体材料的制造。在化学制品、石油、天然气、食品、药物、农药和材料工业的许多制造过程中,高压都是必不可少的。通常有了这种压力才能在化学反应器中获得理想的产率和速率,在分离过程中达到合适的溶剂溶解率,并合成新型高压材料。常见的例子有哈伯法合成氨(压力通常为15~25 MPa);合成气制甲醇(压力通常为5~10 MPa);高压加氢作用、羰基化作用和胺化反应(压力为1.5 MPa);药物合成;超临界气体萃取和反应(压力为7~40 MPa)。在常规工艺中,这些压力是通过压缩体相来实现的。然而,这种压缩是昂贵的,因为其既需要热力学工作,又需要开发能够承受这些压力的多级泵。因此,受到材料的限制,高压工业过程很少使用超过25 MPa(250 bar)的压力。由于施加在容器壁上的压力与被加工的分子所受到的压力相同,更高的压力会导致设备失效。可以在实验室研究更高的压力(使用金刚石砧室使压力高达数百万巴),使得使用新型反应机理[1]合成许多化学品和材料成为可能。然而,采用传统压缩方法的工业生产规模是不切实际的。
在传统的高压工艺中,流体是均匀的,压力是标量常数,在各个方向上都是相等的。压力P=PK+PC,由分子运动(理想气体值PK=RT/V)引起的动力和构型部分PC组成,PC是分子间力和任意外部场作用的结果。除非温度过高,一般构型的影响通常占主导地位(通常是凝聚物质温度下总压力的80%~90%)。这表明,必须通过外场对分子施压来产生高压。这样在流体中会产生不均匀性,因此压力将是一个二阶张量,既取决于力的方向,也取决于它所作用的表面的方向。在本文提出的高压工艺中,固体底物表面施加在吸附相上的相互作用场是尤其令人感兴趣的。
近期有大量的实验证据表明,由于底物提供了很强的吸引力,吸附在固体底物上的流体或固体膜中的分子可以在平行于固体表面的横向上受到强烈的压缩。这种压力是表面驱动的高压效应。此类压缩是通过直接原位实验观察到的,如每个分子占据的底物表面面积的测量[2–5]、X射线衍射(XRD)[6]、低能电子衍射[7–9]、微量热法测量[10]、原子力显微镜和拉曼光谱[11–14]。这些研究中大多数底物表面附近的吸附分子上的压力为1~10 GPa。在分子模拟[15–28]中也观察到了吸附表面层的压缩。在模拟中,底物表面法相压力和平行压力(即切向压力)可单独测量。即使对于只有简单范德华力的小型吸附分子(如惰性气体、氮气和甲烷),切向压力相对于体相压力也有很大提高,前提是吸附物浸湿固体表面;该值取决于底物的性质和与固体壁的距离。对于碳基底,当与吸附层平衡的体相处于0.1 MPa压力时,压力通常为0.5~8 GPa,增强程度为4或5个数量级。应该注意的是,这些切向高压与表面平行,对容器壁本身不施加任何力。法向压力确实对容器壁施加了压力,但要小得多,而且其值既可以是正值也可以是负值[21]。这种压缩对于具有高表面原子密度的固体底物[28]如碳,尤其明显,如图1所示。
《图1》
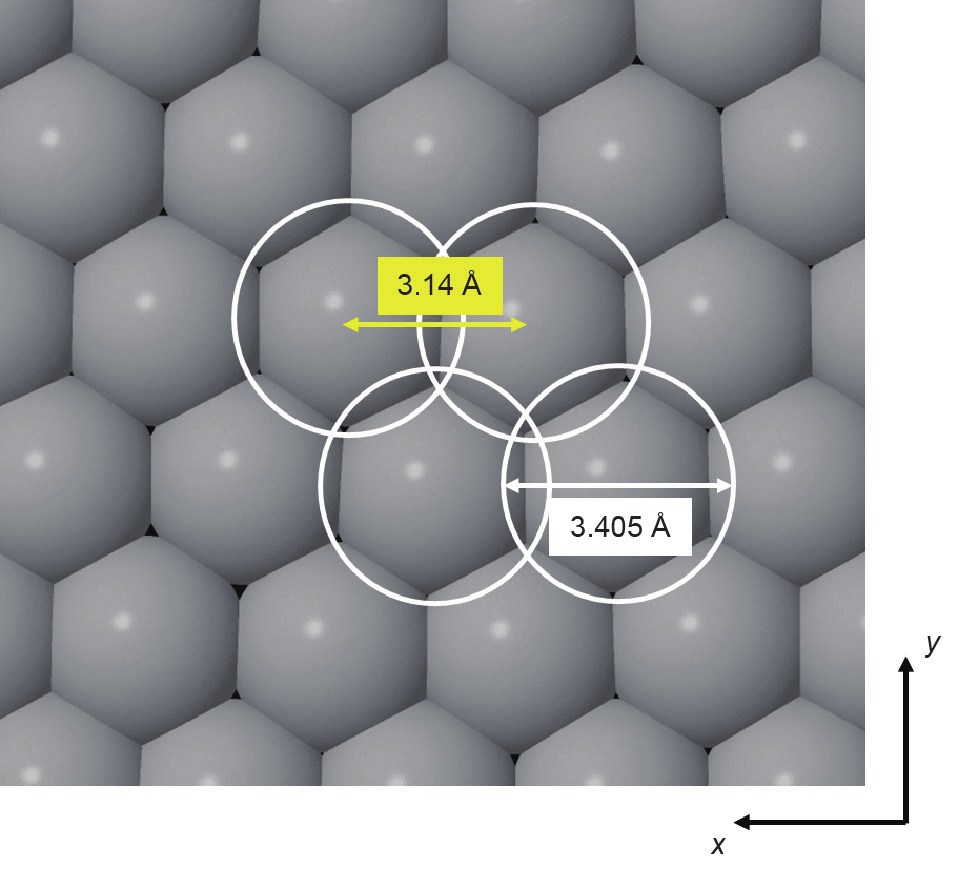
图1.从分子模拟的俯视图来看,位于xy平面内的石墨表面(未示出) 上所吸附的单层Lennard–Jones(LJ)氩(Ar)原子在平行于表面的平面上具有很强的压缩能力。相邻分子(灰色)被3.14 Å分开,它们之间的距离要比当它们之间的偶力为零且不发生压缩(白色圆圈)的情况(3.405 Å)下小得多[28]。
这些最近的研究成果表明,设计高压纳米反应器来合成化学品和原料是有可能的,这些化学品和原料对压力的要求比传统制造工艺要高得多。这些高压发生在底物表面附近。尽管在简单的范德华力的作用下,高压反应器的作用区域仅可以延伸至距表面几纳米的距离(如果存在静电力,则更大),但是由于活性炭通常具有1000~2000 m2·g−1甚至更大的表面积(参见第6节的讨论),总反应体积可能会相对较大。为充分了解这种表面驱动的高压效果,以此来指导这种转型制造方法的设计,我们还需进一步研究。
《2.薄吸附膜的局部压力》
2.薄吸附膜的局部压力
压力是作用在表面元件上的每单位面积的力。在体质均匀的各向同性相中,压力是一个标量,在各个方向和所有位置都是相同的。然而,如果相是不均匀的,如由于固体底物的存在,力以及由此产生的压力,则在不同的方向上是不同的。压力同时也取决于它所作用的表面的方向。因此压力P是一个二阶张量,一般有9个分量;分量Pαβ是在β方向上作用在指向α方向的表面上的每单位面积的力。对于简单几何的底物表面,如平面、圆柱面或球面,只要系统不受应变,压力张量的非对角线元素(如Pxy,即在y方向上作用在指向x方向的表面上的压力)将会消失,仅剩下3个对角线元素。因此,对于平面基片表面或狭缝孔[如图2(a)],其压力张量计算如下式所示:
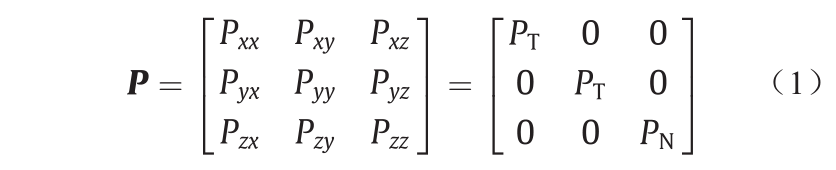
式中,PT=Pxx=Pyy指平行于基片表面的切向压力(xx和yy分量是对称相等的);PN=Pzz是基片表面的法向压力。
流体静力平衡(也称为力学平衡)的条件是:一旦系统达到平衡,在固体底物和吸附相之间的界面上将没有动量的净转移。这导致了以下情形[29,30]:

式中,ρ(r)是分子在r位置的密度(单位体积内的数量),v是作用于r的外场。此处,唯一的外场是重力,而与存在的其他力相比,重力是可以忽略不计的,因此,方程(2)可化为:

方程(3)给出了压力分量的3个条件。对于平面表面,假设表面位于xy平面内,z垂直于表面,条件是PT与x和y无关(如对称性所预期),而法向压力遵从以下条件:
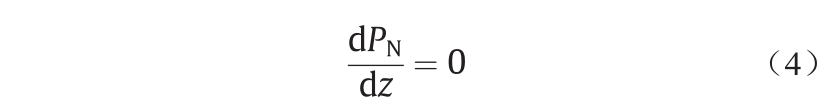
因此,对于给定的系统而言,法向压力是一个常数,与距底物表面的距离z无关。
对于圆柱界面,如圆柱孔[图2(b)],压力张量有3 个对角线分量:一个垂直于孔壁,即PN = Prr,两个平行于孔壁(即切向分量),PTz = Pzz和PTϕ = Pϕϕ,其中(r,z,ϕ)是常用的圆柱极坐标。由流体静力平衡条件[31]得出的方程(3),如图2(b)所示,表明PTϕ和PN是由一阶微分方程联系起来的,而PTϕ 与方位角ϕ无关,且PTz与z轴无关。后两个条件是由对称性推出的。将ϕ方向的法向压力与切向压力联系起来的一阶方程表明,在圆柱几何 形状中实际上只有两个独立的压力,即PN和PTz。
《图2》
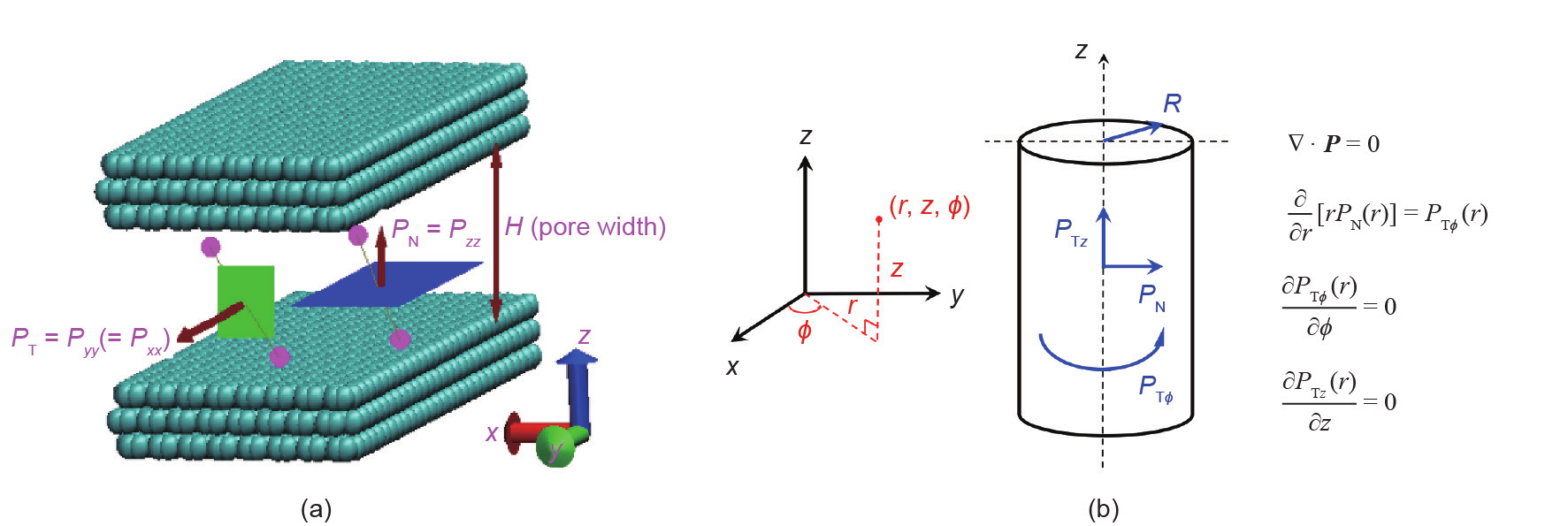
图2.(a)为一种每壁有3层石墨烯狭缝状的碳孔,展示了法向压力PN、切向压力PT和它们作用的表面;(b)为圆柱形孔,给出了法向压力、两切向压力和流体静力平衡方程。
《3.定义非均匀相中的有效压力》
3.定义非均匀相中的有效压力
计算限定纳米相中r点处局部压力的常规途径是所谓的维里或力学路线,在此过程中,我们将作用于表面 元素的分子间作用力加总并且除以区域面积[29,30]。至于对加电位(pair additive potential),如下所示:

式中,u(ij)是分子i和j之间的对势;Cij是连接分子i的质量中心ri到分子j的质量中心rj的任意等高线,其中rij=rj–ri;kB为玻尔兹曼常量;T为温度;1为单位张量;δ(·)为狄拉克三角函数;<∙>表示系综平均。
由于分子的动能,方程(5)右端的第一项是理想气体压力,而右侧的第二项是分子间力引起的压力的一部分。r点的压力在一定程度上取决于等高线Cij的选择,而Irving和Kirkwood(IK)的压力是常见的选择[32]。在IK选择下,等高线是连接分子i和j中心的直线,如果这条线与区域的元素相交,力就会对压力起作用。这种在纳米尺度压力定义中的不唯一性是因为分子对(或三重态等)之间的作用力,并且还不存在一种特殊的方法将它们分配至空间中的某一特定点r。在计算或测量纳米尺度的压力时,有必要像IK所做的那样,对P进行操作定义。我们可以把这样的压力看作是作用于某一特定方向的近似有效压力。当在适当的距离上进行均分时,这种局部有效压力应能给出定义明确的唯一值,而并不依赖于所使用的操作定义[29,30]。值得注意的是,在平面表面的特殊情况下,即使切向压力不是唯一定义的,而法向压力却是唯一定义的。这是从方程(5)得出的,这意味着底物表面附近的PN值与远离表面的PN值是相同的。
热力学路径是维里路径的一种替代方案[25],这种方法通过微小扰动改变体积ΔV,长度在α方向上受到干扰,其他维度Lβ保持不变。压力分量Pαα如下:

式中,A是亥姆霍兹能;ΔU是由于体积微小扰动产生的组态能变化,并且总体平均能量超过未扰系统。热力学路径相比维里路径的优势在于利用热力学路径在力场中能更容易涵盖三体和产生更高效应。
除了通过维里或热力学路径得到P外,在一些情况下,还能通过第三种方法求得压力,而这种方法基于将吸附层结构与二维(2D)或一维(1D)系统的状态方程(EOS)相结合,分别应用于狭缝孔和圆柱孔。这种方法的优点在于,由于2D和1D系统是均匀系统,其压力明确可得,此外,在任意三维(3D)均匀系统中也同样如此。我们已经在2D系统中对这种方法进行了初步测试。靠近孔壁的吸附层是狭缝孔的准2D模型(图3)。在模拟过程中,可以将给定层上分子的中心位置投射到规定的2D平面,同时记录2D密度。如果状态方程可以运用于2D系统,那么可得到与2D密度相对应的2D压力。通过将2D压力除以适当长度,如密度半峰宽,上述方法能运用于估算3D狭缝孔系统的有效切向压力[33]。由于存在压缩现象,这些吸附层的2D密度极高,是熔点处密度的两倍。在这种高密度下,我们为Lennard-Jones(LJ)系统研究了一种高准确度的2D状态方程[33],并用它来估算有效切向压力从而得到大范围的润湿参数和孔缝宽度。通过“2D路径”和维里路径得到的PT值极其一致,通常差别在10%~20%之间[见图3(a)]。
《图3》
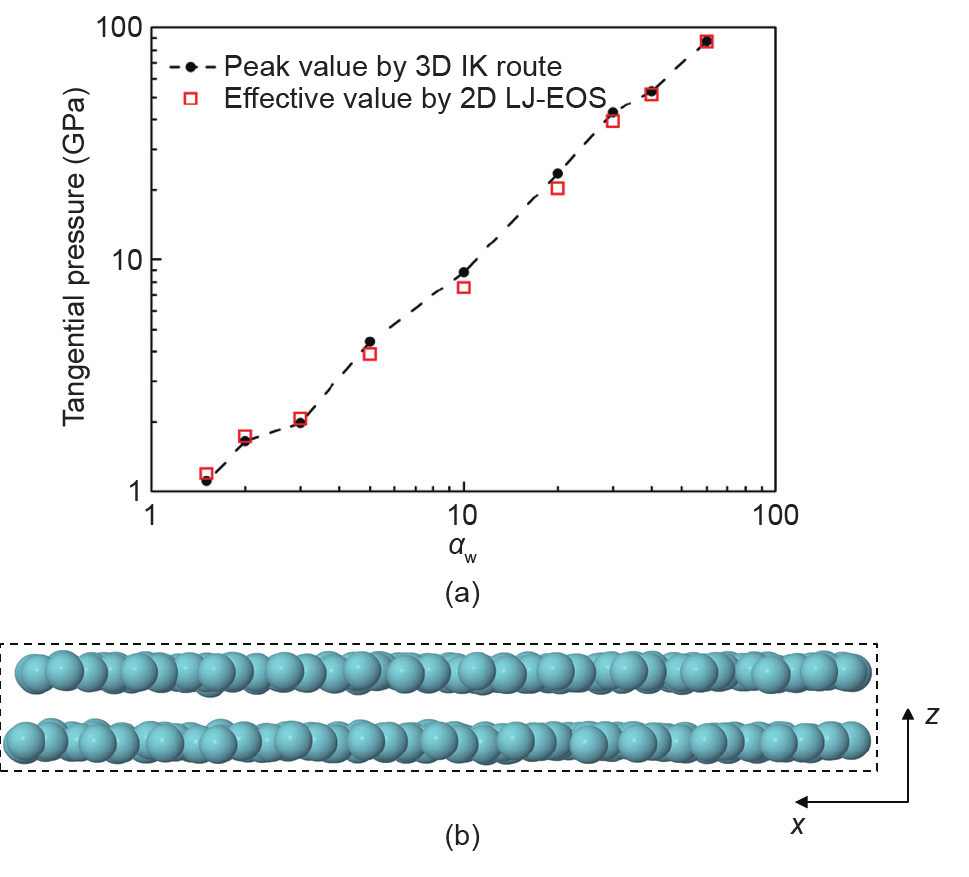
图3.(a)在3个分子直径宽度的狭缝孔中处于Ar的沸点(87.3 K,1 bar体相压力)时传统3D IK路径和2D路径求得LJ Ar切向压力的对比。其中,αw微观润湿参数(定义见第5节);(b)狭缝孔中两个吸附层的正视图,3个分子直径的宽度为αw=10。
《4.表面驱动高压的实验证明》
4.表面驱动高压的实验证明
迄今为止,直接测量固体底物表面附近压力的实验技术尚未开发。而作用于底物表面的法向压力PN有可能测得,因为PN能引起底物材料结构变化(如孔缝宽度或层间距的变化),这种变化可通过XRD或中子衍射观测到[21,34]。对于平面而言,PN是与到平面距离z无关的常数,但对于曲面而言这种说法不成立(参照图2中圆柱面的流体静力学平衡状态)。在分子占据面积的吸附量测试[2–5]、微量热法测量[10]、XRD[6]和低能电子衍射[7–9]中能在原位直接观察到固体表面附近分子产生的压缩现象。对于具有高表面原子密度的固体底物而言,如碳,这种压缩效应最为明显。
除了上述直接观察固体表面附近的分子压缩现象外,还有很多证据间接表明这种表面膜上存在高压。实验研究表明,在纳米孔中能形成高压相,以及在纳米孔或其表面上进行化学反应产率很高,而当反应在体相中进行时,为取得这种产率通常需要极高的压力。下面将讨论一些这种类型的相关例子。
《4.1.纳米孔中形成的高压相》
4.1.纳米孔中形成的高压相
尽管与孔相平衡的体相处于常压或常压之下的状态,在纳米多孔材料中仍常能形成高压相[35–41]。例如,体相碘化钾(KI)在常温常压下形成B1 NaCl型晶体结构( 空间群);而在1.9 GPa压力下,将会转变成B2 CsCl型晶体结构(Pm3m空间群)[42]。然而,Urita等[35]通过高分辨投射电子显微镜(HRTEM)和同步XRD表明,当被限定在内径为2~5 nm的单壁碳纳米管(SWCNT)时,即便与孔相平衡的体相KI处在低于0.1 MPa的压力下,还是能形成高压B2相。另一个例子是,在碳纳米孔中能形成甲烷水合物[43],而这原本是一种在体相中需要高压才能发生的相变。除了在低体积压力下形成水合物,碳纳米孔的限制使得水合物形成在动力学上显著增加。
空间群);而在1.9 GPa压力下,将会转变成B2 CsCl型晶体结构(Pm3m空间群)[42]。然而,Urita等[35]通过高分辨投射电子显微镜(HRTEM)和同步XRD表明,当被限定在内径为2~5 nm的单壁碳纳米管(SWCNT)时,即便与孔相平衡的体相KI处在低于0.1 MPa的压力下,还是能形成高压B2相。另一个例子是,在碳纳米孔中能形成甲烷水合物[43],而这原本是一种在体相中需要高压才能发生的相变。除了在低体积压力下形成水合物,碳纳米孔的限制使得水合物形成在动力学上显著增加。
通过表面力仪实验观察到有机化合物的固液转变通常发生在远高于其正常熔点Tmp的温度,这也意味着其正处于高压状态。在这些实验中,有机纳米相限制于云母表面。例如,正十二烷(Tmp=263.4 K)在300 K时凝固,而环己烷(Tmp=279 K)在296 K于云母表面上凝固[37,39]。在体相中,正十二烷在300 K、约1860 bar的状态下凝固,而环己烷在约440 bar的压力下凝固。这些受限的高压效应与分子模拟研究相符,其中与受限纳米相平衡的体相,其压力相对较小的增加都能导致纳米孔中的切向压力的大幅增加[20,24,25]。
《4.2.受限纳米相分子结构的变形》
4.2.受限纳米相分子结构的变形
2D石墨烯的发现引起人们对2D材料的浓厚兴趣,目前约有1000种2D材料已被合成或预测可合成,包括2D硼和其他氮化物、2D钼硫化物和2D过渡金属硫属化物[44,45]。最近,研究报告了各种纳米相被限制在平行2D薄板组成的狭缝状孔隙中,这些受限的分子受到了高压[46,47]。Vasu等[12]研究了被限制在两片狭缝中的苯胺和硼,这两片狭缝由石墨烯组成,孔径宽度为1 nm。这些分子结构对压力很敏感,研究人员使用拉曼光谱来确定石墨烯孔隙中分子的结构。通过比较观察到的分子结构和拉曼研究块状材料得到的结构,他们能够在孔隙中估测到一个1.2 GPa的有效压力。这一发现与我们获得的狭缝炭孔的分子模拟结果吻合[20,25]。最近这个研究小组和其他小组在其他相似的实验中观察到了相似的高效压力,范围为1~10 GPa[11,13,14,46,47]。虽然这些实验证实了碳孔中强压力有所增强,但这一压力并没有很好地被定义,因为“有效的体积压力”是标量,而孔隙中的实际压力是二阶张量。
《4.3.纳米孔隙中的高压反应》
4.3.纳米孔隙中的高压反应
发生于体相中的化学反应需要高压的存在,即使是在与孔隙相平衡的体相处于低压情况下,这些反应在纳米多孔材料中进行时往往会达到高产。这里我们举两个例子。
4.3.1.一氧化氮二聚化反应
1989年,Kaneko等[48]报道了一项研究,这一研究是有关发生在温度为273~423 K,狭缝状活性炭纤维(孔径为0.8~0.9 nm)孔隙的一氧化氮(NO)二聚体反应,2NO=(NO)2。磁化率测量结果表明,即使在与孔隙平衡的体相中二聚体的摩尔分数很小,纳米孔隙中的反应也能基本完成(超过98 mol%二聚体)。Byl等[49]后来的实验证实了这一令人惊讶的结果,他们研究了发生在(10,10)SWCNT(直径为1.36 nm)、温度范围为103~136 K的反应。用傅里叶转换红外光谱仪(FTIR)测定了SWCNT中的平衡成分,在实验误差范围内,二聚体的摩尔分数为100%。随后由不同的组研究团队[50–52]进行的理论和分子模拟(反应性蒙特卡罗)未能重现实验结果。二聚体的预测产量虽然高于体相,但比实验中发现的要低得多。预测结果表明,随着温度的升高,产量也会迅速下降,而实验结果显示温度对产量的影响不明显。最近,Srivastava等[53]的研究表明,早期理论预测的失败是由于忽视了碳壁中二聚体和π电子之间的强缔合相互作用。从MP2(二阶Møller-Plesset微扰理论)阶段“从头”(Ab initio)计算表明(NO)2二聚体分子与碳壁之间的相互作用力比范德华力所预期的强20倍,在先前研究中也曾设想过这一结果。考虑到这一点,在分子模拟中,活性炭纤维和碳纳米管的产量和温度依赖性在实验中有良好的一致性。由于孔壁上的强吸引力,在孔壁附近的z方向的切向压力(平行于孔壁)以数十万巴的单位被计算。
4.3.2.水溶液中的反应
Vasu等[12]研究了石墨烯狭缝孔隙对氯化镁(MgCl2)、碘化铯(CsI)、硫酸铜(CuSO4)和氢氧化钙[Ca(OH)2]几种盐类水溶液的影响。他们发现在室温下这些化合物与水在体相条件下未发生反应。由于纳米孔隙中的高压,这些化合物与水在密闭的纳米相产生相应的金属氧化物(除CsI以外)。他们使用能量色散X射线(EDX)光谱、电子衍射和电子能量损失能谱(EELS)证实了氧化物的转化。这些实验明确地证实,在孔隙中观察到的晶体是氧化物。
由本体水溶液[54]中的高压实验可得知,压力从0.1 MPa(1 bar)增加到1 GPa(10 000 bar)导致水的电离常数增加两个数量级,从而促使盐水解。研究人员提出,密闭诱导的高压导致盐水解到其氢氧化物后分解为相应的氧化物。在体相中,这些盐类物质在他们的氢氧化物或氧化物发生转换之前,只有在高温和高压下才能被观察到[55]。因此,纳米限制及其产生的高压足以让反应在室温下进行。他们还研究了在这些石墨烯孔隙中溶解于甲醇的MgCl2,并没观察到反应发生,因为没有电离的溶剂,所以这一发现是符合预期的。Lim等[46,47]最近报道了其他有关水在纳米密闭下的高反应性的实验观察。
《4.4.约束条件下的新型高压材料合成——绝缘体–金属转变》
4.4.约束条件下的新型高压材料合成——绝缘体–金属转变
Fujimor等[56]报道了硫受限于SWCNT或直径约1 nm的双壁纳米碳管(DWCNT)中并与大量负压下的硫蒸气处于平衡状态时,形成了1D金属硫相[图4(a)]。HRTEM和同步加速器XRD的观测结果可以证实,受限的硫形成线性还是锯齿形共价链取决于纳米管的精确直径。这些链有较长的结构尺寸(可达160 nm),并在温度约为800 K时仍具有热稳定性。线性链的S—S键长约为0.19 nm,比键长约为0.207 nm的体相聚合硫SΨ的键长短,这表明线性链的S—S有很强的化学键合。减少的键长和1D相的金属性质表明硫链在孔内具有很强的周向压力。这种受限硫的特性与自由硫的特性形成了强烈的对比,因为1D链不稳定且3D金属相只在约95 GPa的压力下形成。研究小组[27]最近进行的分子动力学(MD)模拟研究预测硫链中心的周向压力约为700 GPa,这远高于金属硫形成时的体相压力。
《图4》
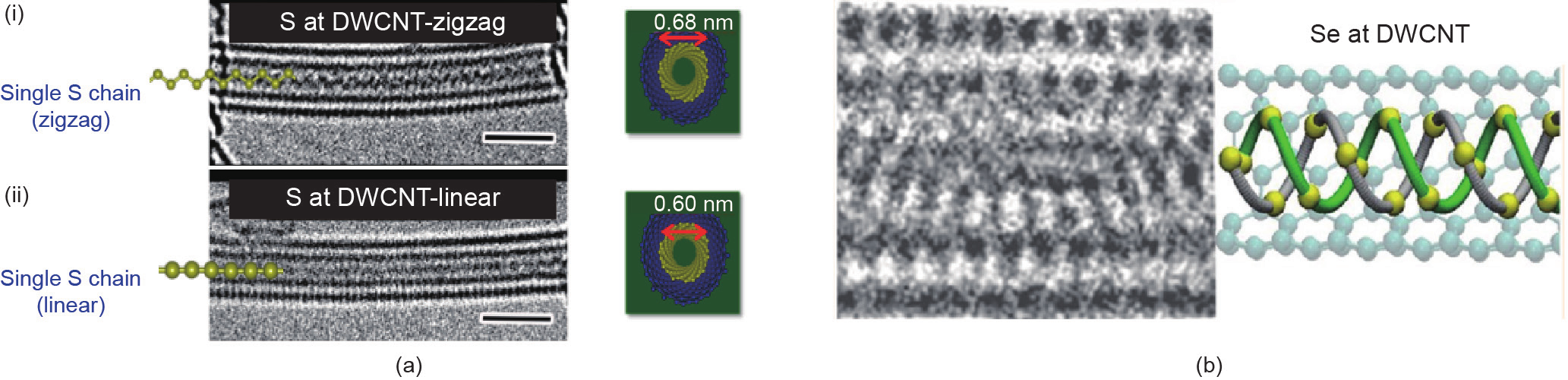
图4.(a)被限制在纳米碳管内的硫(S)的HRTEM图像:(i)在直径为0.68 nm的DWCNT内的锯齿形链;(ii)在直径为0.60 nm的DWCNT中的1D硫线性链[56],比例尺为1 nm;(b)被限制在直径约为1 nm的DWCNT中的硒(Se)的双螺旋结构[57](改编自文献[56,57]) 。
Fujimori等[57]也观察到了在直径约为1 nm的DWCNT中键合型硒(Se)的双螺旋结构[图4(b)]的形成,HRTEM、XRD和“从头”(ab initio)计算的结果可以证实。受限的双螺旋结构Se在孔中被径向压缩。这是Se的一个新的结构相,与自由Se的结构截然不同。
最近,Medeiros等[58]报道了包裹于直径位0.7~1.1 nm的SWCNT的1D碲链的形成。随着纳米管直径的增加,能观察到从金属特性向绝缘特性的转变。纳米限制不仅是生成这种新型相的手段,而且对于确保封闭物的力学稳定性也是必要的。
《5.表面驱动高压的理论和分子模拟研究》
5.表面驱动高压的理论和分子模拟研究
对于与简单范德华力相互作用的非反应性系统,有简单几何形状的纳米孔中的吸附薄膜的法向压力和切向压力已经得到了相当广泛的分子模拟研究。对于发生化学反应的情况,无论是在吸附薄膜中的分子之间,还是在这些分子与孔壁之间,我们只做了少量研究,且目前这些系统中的压力情况尚不清晰。
《5.1.具有简单范德华力的非反应系统》
5.1.具有简单范德华力的非反应系统
从2011年开始,许多研究使用半巨正则蒙特卡罗(SGMC)模拟来计算在纳米多孔碳和具有狭缝、柱状和球状孔的硅中的简单非极性流体的压力张量分量[20–22,24–28]。这些研究证明,法向压力和切向压力分量通常非常大——法向压力高达数千巴,切向压力高达数万巴。这些压力值与至今为止的实验观察结果定性相符。在模拟中,孔隙的长度是有限的,且与大气直接接触,从而确保受力(或静压)平衡(即平衡时无净动量转移),方程(3)符合此条件。
图5(a)显示,在87.3 K、1 bar体压(Ar的沸点)和表现出简单范德华力的吸附分子(LJ)的相互作用条件下,可以得到碳孔的宽度为15分子直径(约5 nm)这一典型结果。邻近孔壁的接触层中的切向压力约为19 000 bar,而在孔的中心压力在约200 bar的位置振荡[见图5(a)中的插图,其显示了膨胀时的压力]。对于较小的孔隙,PT要高得多——对于在液相区温度范围内宽度为两个分子直径的小狭缝纳米孔,PT高达60 000 bar[20,25]。这些巨大的正切向压力来自于xy平面上相邻分子间的强排斥力(图1)。在系统中显示为黑色水平线的法向压力PN在大约27 bar的孔隙中是恒定的。图5(b)显示了实验和模拟中限制于狭缝孔隙(一个活性炭纤维模型)的四氯化碳(CCl4)的法向压力及孔径。通过XRD数据观察石墨烯层的层间距变化以及估计的杨氏模量获得实验值。法向压力可以是正值也可以是负值,这取决于孔的宽度。通过各项试验研究可得知,随着孔隙宽度增加,且受到孔隙包裹作用的影响,可能导致这种振荡的发生。
《图5》

图5.(a)(左)Ar在正常沸点下空隙宽度为H=15σAr的裂隙状碳中的法向压力和切向压力[25];(右)模拟气相平衡时的纳米孔相的快照;(b)温度在300 K时活性炭纤维中吸附CCl4的法向压力在不同孔径(H)下的变化[21](改编自文献[21,25])。
通过对压力的统计力学方程的检验可以发现,压力取决于温度、体相压力以及固体表面上的吸附分子的润湿特性。润湿程度的定量测量由微观润湿参数αw[20]所确定,αw即流-固耦合与流体-流体相互作用的比率,如下式所示:

式中,ρs和Δ是固体原子的数密度以及固体中原子层之间的间距(石墨ρs=114 nm−3,Δ=0.335 nm);ε和σ是流-流(吸附物-被吸附物)作用(ff)和流-固耦合(fw)的LJ参数。分子模拟研究表明,随着润湿参数的增加,切向压力迅速增加(图3)。与孔相平衡时,它也会随体相压力的增加而迅速增加(大致呈指数型)。
《5.2.发生化学吸附或化学反应的受限系统》
5.2.发生化学吸附或化学反应的受限系统
虽然在发生化学吸附或化学反应时,几乎没有关于应用于薄吸附膜的表面驱动高压的研究报告,但至少在某些情况下,切向压力可能会变高。最近,Srivastava等[28]报道了一项系统研究,该研究涉及表面润湿程度、孔隙宽度和吸附分子形态对裂隙状孔隙压力张量分量PN和PT的影响。本研究包括被孔壁弱化吸附的分子,以及在αw值高达40的情况下,一氧化氮二聚体(NO)2与碳壁的化学吸附。在这些较强的相互作用中,孔壁附近的切向压力在1×105~5×105 bar之间。
最近,有报道指出MD模拟[27]在碳纳米管(CNT)中具有与Fujimori等[56]的实验中所使用的相同直径和手性的硫[见图4(a)和相关讨论]。Stillinger和Weber(SW)反应力场[59]的一种改进形式被用于硫相互作用。封闭硫纳米相的结构和切向压力,如图6(a)和(b)所示。强共价结合的硫原子,键长为0.189 nm,与(0.18±0.02)nm的实验值较为吻合[27],但在体相上远小于聚合硫SΨ,其键长为0.207 nm。局部切向压力的峰值约为700 GPa。如实验所观察到的,这种高压足以产生金属相(本体硫的绝缘体金属相变发生在95 GPa左右)。这些计算本质上是定性的,因为所使用的力场适用于体相二聚体属性。然而,有实验证据表明,封闭纳米相的结合强度接近于本体二聚体[27]。在MD模拟中,将力场能量改变10%~20%导致计算出的切向压力发生变化,但压力还保持在4~10 GPa的范围内,这足以解释硫链的金属行为。
对孔隙中硫原子所经受的力和势能的分析表明,其与孔壁的相互作用是强烈排斥的[图6(c)]。然而,硫-硫相互作用是有强吸引力的,因为通过化学键合,硫链可以通过进入微孔降低其自由能并且被碳纳米孔稳定住。对切向压力原因的检验表明,正是由于强硫-硫键造成的这种与孔壁的相互排斥作用,产生了巨大的压力。
《图6》
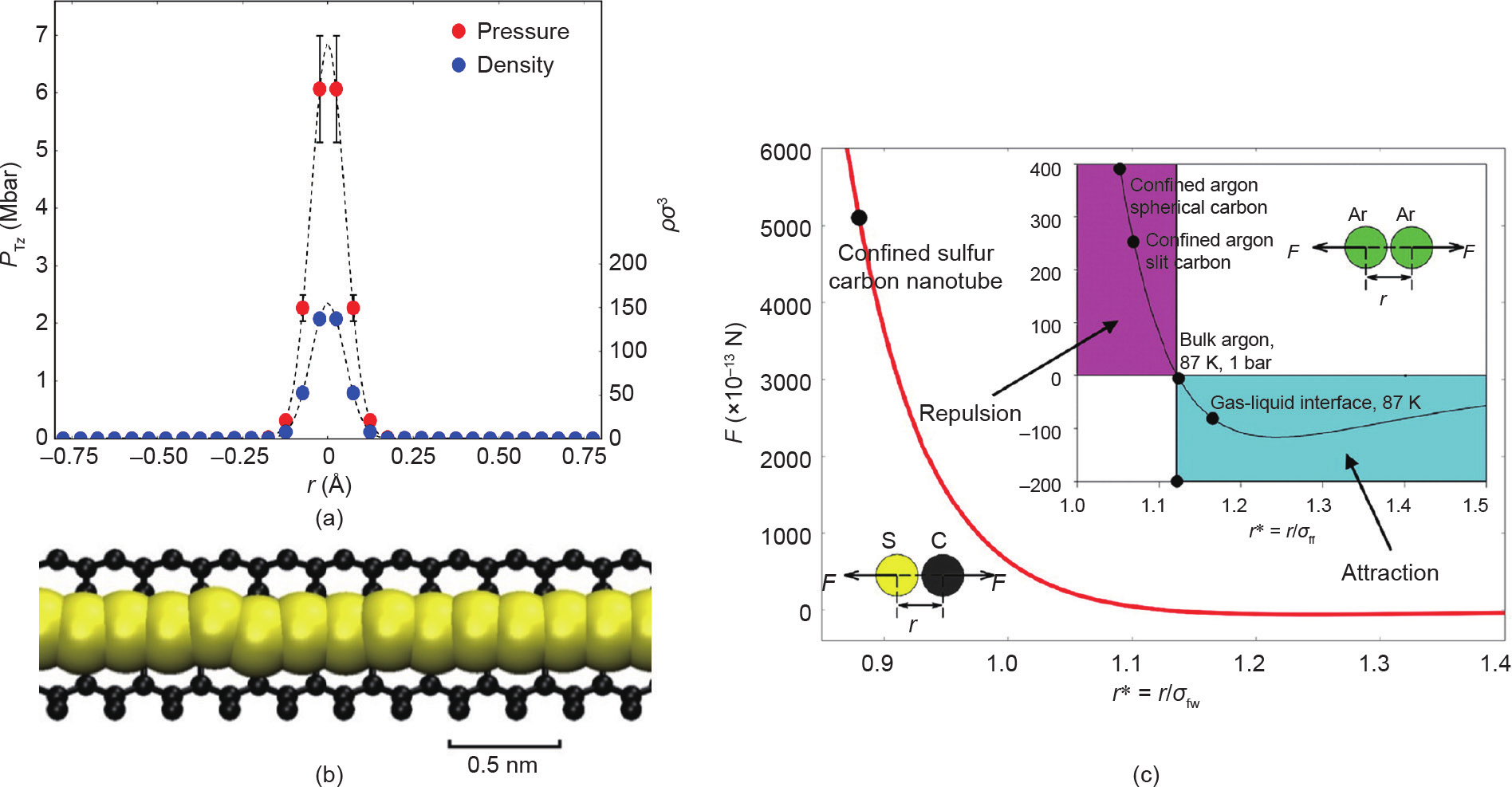
图6.(a)硫原子的切向压力和数密度;(b)MD模拟中的直径为0.626 nm的(8,0)SWCNT中的1D硫;(c)对孔隙中硫原子所经受的力和势能的分析:红色曲线表示(8,0)SWCNT中硫的硫原子(S)和碳壁原子(C)之间的分子间作用力;黑色圆圈表示平均硫-碳间距的力。插图是狭缝形和球形碳孔中碳壁附近的LJ Ar原子的相应Ar/Ar力曲线[27](改编自文献[27])。
《6.结论》
6.结论
压力是一种基本热力学变量,因其具有大量理想的特性,而有可能控制和促进化学物品与固体材料的制备。现有的工业过程通过机械压缩来实现高压,典型的如大宗混合气体的压缩,这一点已被Fritz Haber证明——1909年,他以20 MPa左右的压力,通过氢气和氮气生产出了氨。然而,由于成本高昂和压缩机设计困难(即材料方面的考虑),这种高压工艺在应用上受到限制。表面驱动的高压化学反应、化学加工和材料合成的发展将带来巨大的效益,并能将压力的使用变成工业制造中的决定性变量。由于高压是由底物表面的引力场提供的,这就避免了传统压缩的高成本和材料限制。更重要的是,这种表面驱动的高压工艺提供了一种简单的方法,可以达到比目前工业规模更高的压力,高达上万巴,在某些操作中甚至更高。此外,由于主压力分量是平行作用于固体表面的切向压力,因此可以避免过高的压力作用于反应堆容器壁,从而避免材料的失效。
首先人们可能会对几种针对表面驱动高压效应的实际应用提出异议:
(1)由于高压区仅在底物表面延伸几纳米,所以活性反应区将非常小。然而,简单的计算表明,通常情况下并非如此,主要是因为活性炭具有非常大的表面积——通常为1000~2000 m2·g–1。这种碳具有约0.5 g·cm–³的密度和1~2 nm的微孔宽度。假设碳的表面积为1000 m2·g–1,平均孔径为2 nm(在活性反应区),那么直径2 m、高10 m的吸附柱将含有约15 000 kg的碳和一个30 m3的活性反应区。
(2)离开活性反应区后,压力较低,生成物恢复到原来的低压状态。这只有在过渡壁垒较低且产物在低压条件下处于非平衡相的情况下才会发生。但是,该现象在大多数情况下是不会发生的,因为最终产物要么处于
精确的热力学平衡状态,要么处于亚稳状态但由于高活性势垒(如一般条件下的金刚石)而无法恢复真正平衡。例如,在实验室里,由于高活化能势垒,许多化学反应受到动力学限制,因此需要高压(通常为0.1~3 GPa)来克服这一问题。
(3)有些应用可能会出现难以将产物从微孔中移出的情况,如在窄碳纳米管中生成硫和碲的纳米线。但是,在很多应用中移出生成物是有可能的,如溶剂萃取、超声、降低体积相压力或其他工艺。
《7.展望》
7.展望
为了在实践中运用表面驱动的高压工艺,首先需要 建立完备的基础知识库,包括生产、控制和测量这种表 面驱动高压的最佳方法。要获取建立这些进程的理论模 型的可靠方法,就需要开发和测试电子、原子和中等尺 度物质的方法,以便解释约束和强压缩,并确定反应机 制,活跃状态和阻碍,反应速率和扩散率,这些都在限 制之内。基础研究也需要开发实验方法,以便更精确地 控制和表征纳米多孔材料合成中的表面几何形状、化学 性质、孔径大小和形状,并研究和控制反应物分子进入 孔隙及物质的排出。
这一领域尚处于起步阶段,许多方面有待进一步研 究。包括:
(1)研究需要高压的化学反应原型。在一些情况下,表面驱动高压可能有助于由平衡热力学引起的反应的发生,如由于发生反应而使得摩尔数减少(勒夏特列原理)。在这些情况下,反应产率的实验研究应与分子模拟结合以计算平衡产率(反应性蒙特卡罗)和自由能(如通过直方图重新加权或密度状态方法[60])。在由于高活化屏障而受动力学限制的反应中,至关重要的是研究克服屏障必须具备的压力以及如何通过操纵吸附材料、孔径和形状来控制反应。我们是否可以利用固体底物的表面特性来选择最佳反应途径和最终产品物?此外,应该将实验调查与理论(例如,密度泛函理论和串优化方法以确定反应路径和活化状态)相结合。
(2)水溶液中的反应。在体相中的水和水溶液的高压实验表明,水的电离常数按照数量级增加[54],这导致反应性大大增加[55]。最近,已有几项研究[12,46,47]观察到在石墨烯纳米气泡内的盐溶液中有水解反应,这显然是由表面驱动压力造成的。该领域涉及广泛,有待进一步研究调查。
(3)表面驱动的高压对相变的影响。纳米孔和固体底物上的纯物质的气液冷凝和熔融/冷冻方面已经有了大量的研究。然而,诸如蒸馏、气体吸附、液-液萃取、膜分离、结晶和超临界萃取等工业过程常掺杂混合物,人们对于混合物的表面压缩效应知之甚少。许多领域的研究过少甚至尚未开展,包括表面驱动压缩和限制对稀溶质在液体溶剂中的溶解度的影响[61]、对在6种已知的二元混合物[30,61–64]相图的流体和固体区域上的影响以及对高压固-固转变的影响[62,64]。
(4)探索生产药物和其他特种化学品所需多晶型物(包括新多晶型物)所需的条件。这些条件可能包括吸附底物的类型、表面条件、孔径大小和形状。
(5)超高压对材料电子和光子特性的影响[1]。格外引人注目的是由高压引起的电子带宽和带隙的变化,以及由此产生的绝缘体-金属、绝缘体-半导体和半导体-金属转变以及向磁和超导状态的转变[1,65–69]。研究人员使用冲击压缩或砧室测量,已经对自由元素和化合物的这些转变做了大量研究。在自由物质中,这些转变发生在低于1 GPa至200 GPa左右的压力下。最近的研究表明,在SWCNT中限制合适的直径足以引起硫[56]和碲[58](两者都是周期表中的VIB族元素)的绝缘体-金属转变,并且理论计算[27]表明这是一个表面驱动的高压效应,切向压力为几百吉帕(图6)。
(6)表面驱动高压对原子和分子性质-分子构型、振动模式、键角等的影响。这些研究的方法可能包括FTIR和拉曼光谱学[12]。
(7)测量固体底物附近的吸附纳米相的压缩实验技术的发展以及这些相的局部性质,包括底物表面附近的有效切向压力。通过使用小角度中子衍射来确定局部密度以及切向压力的理论EOS,可以估算后者在平面上的吸附单层(参见图3和相关讨论,其中有效切向压力是根据压缩吸附单层中的数密度的分子模拟结果估计的)。FTIR和拉曼光谱为观察表面附近压缩对单个分子的影响提供了一种可行的途径。
《Acknowledgments》
Acknowledgments
The authors would like to thank several colleagues for helpful discussions and cooperation on the topic of pres- sure enhancement in thin adsorbed films, including Cody Addington, Katsumi Kaneko, Jeremy Palmer, and Fanxing Li. We also thank two anonymous referees for helpful comments and suggestions. We are grateful to the US National Science Foundation (CBET-1603851 and CHE-1710102) for support of this work. Małgorzata S´ liwin´ skaBartkowiak thanks the National Science Center of Poland(- DEC-2013/09/B/ST4/03711) for support.
《Compliance with ethics guidelines》
Compliance with ethics guidelines
Keith E. Gubbins, Kai Gu, Liangliang Huang, Yun Long, J. Matthew Mansell, Erik E. Santiso, Kaihang Shi, Małgorzata S´ liwin´ ska-Bartkowiak, and Deepti Srivasta- va declare that they have no conflict of interest or financial conflicts to disclose.











 京公网安备 11010502051620号
京公网安备 11010502051620号




