

《1. 引言》
1. 引言
自从1796年和1928年分别发现和开发第一种疫苗和第一种抗生素以来,天花或霍乱等传染病导致的死亡率显著下降。发达国家的平均人口预期寿命从47岁大幅提高到80岁[ 1–3 ]。因此,癌症作为困扰世界的最具灾难性的疾病之一,将其攻克已经成为重中之重。在美国这个拥有一些最先进的癌症治疗疗法的国家,癌症仍然是第二大死亡原因。2015年,癌症夺走了595 930人的生命,仅比心脏病死亡人数少37 912人[4]。尽管在尼克松总统宣布对癌症宣战前后,癌症治疗取得了进展,包括手术、放射治疗、化疗和最近的靶向治疗,但不管癌症患者的生活质量如何,只有大约44%和28%的癌症患者分别活了10年以上和15年以上[5]。
最近开发的一些新的治疗方法为改善癌症预后提供了可能性,这些治疗方法包括免疫调节和工程化重定向细胞治疗(图1)。临床试验NCT00924326的相关数据显示,自体抗CD19嵌合抗原受体T细胞(CART19)能够诱导患者晚期滤泡性淋巴瘤消退。与临床结果平行,治疗后至少39周,B系细胞因CART19治疗得到根除[6]。2011年,我们宾夕法尼亚大学的研究团队和同仁发表了两篇论文,讲述了自体CART19疗法成功治疗复发难治性慢性淋巴细胞白血病(R/R CLL)患者[ 7,8 ]。相关报道首次证明了靶向CD19恶性肿瘤的工程化T细胞对人类有持久的治疗效果。
《图1》
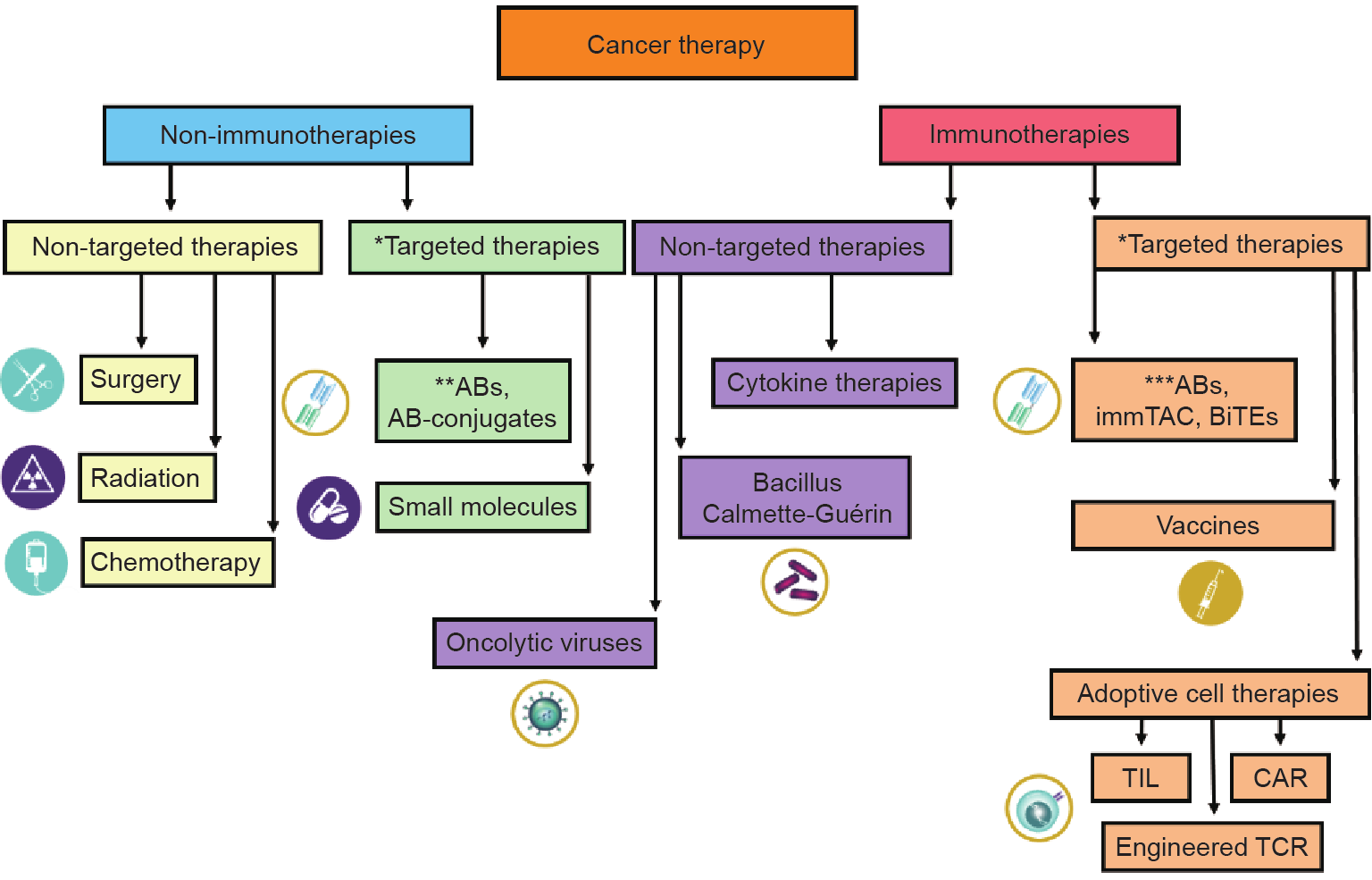
图1. 根据作用机制对各种癌症疗法进行分类。*:通过靶向肿瘤特异性基因起到的治疗作用;**:与癌细胞表面靶结合并导致癌细胞死亡的单克隆抗体(mAB),如曲妥珠单抗;***:检查点抑制剂,如派姆单抗;AB:抗体;ImmTAC:免疫动员抗肿瘤单克隆T细胞受体;BiTE:双特异性T细胞衔接子;TIL:肿瘤浸润淋巴细胞;CAR:嵌合抗原受体;TCR:T细胞受体。
为了清楚地定义和区分CART19疗法与其他治疗方式,图1显示了根据药物靶标作用机制分类的当前癌症疗法的示意图。癌症疗法可分为非免疫疗法和免疫疗法。免疫疗法是直接调节患者自身免疫系统以取得有益临床效果的治疗方法[9]。与免疫疗法相比,所有其他癌症疗法均属于非免疫疗法。溶瘤病毒(oncolytic virus,OV)治疗同时具有两组疗法的特点,因为该疗法直接杀死细胞并诱导抗肿瘤免疫。因此,Kaufman等[10]将溶瘤病毒治疗描述为一种新型免疫治疗药物。每组疗法可进一步分为靶向治疗和非靶向治疗。靶向治疗是在分子水平上根据癌症特异性靶标开发的治疗,靶标结合导致特异性癌细胞死亡[ 11,12 ]。与靶向治疗相比,所有其他疗法均属于非靶向治疗组。
靶向免疫疗法包含一个被称为过继细胞疗法(adoptive cell therapy,ACT)的分组,由活体外扩增的未修饰肿瘤浸润淋巴细胞(tumor-infiltrating lymphocyte,TIL)、工程化T细胞受体(T cell receptor,TCR)和嵌合抗原受体(chimeric antigen receptor,CAR)T细胞组成[13]。癌症过继T细胞转移疗法旨在提高T细胞对抗癌细胞的活性。靶向CD19的CART19疗法由工程化自体/同种异体T细胞组成,特异性定向该细胞使其靶向大多数B细胞上表达的CD19蛋白。这些T细胞经过“训练”,能识别并杀死B细胞[8]。在最近对20项已发表的CAR T细胞临床试验的综述中,11项试验属于CART19试验。这11项试验中,有10项证明了B细胞恶性肿瘤患者得到一些临床益处[14]。美国食品药品监督管理局(Food and Drug Administration,FDA)根据来自NCT02435849的数据批准这种疗法用于难治性或继发性或复发前体B细胞急性淋巴细胞白血病(acute lymphoblastic leukemia,ALL)患者(25岁以下)的治疗。此疗法目前称为Kymriah(tisagenlecleucel)[15]。此后不久,FDA根据来自NCT02348216的数据批准Yescarta(axicabtagene ciloleucel)用于治疗至少接受过两种其他治疗方案后无缓解或复发的特定类型大B细胞淋巴瘤成年患者[16]。随后,根据来自NCT02030834的数据,Kymriah经批准用于治疗大B细胞淋巴瘤[17]。CART19疗法的成功证明了工程化T细胞疗法的理念。这也标志着现代医学新领域的开始,即工程化T细胞疗法。该疗法可定向攻击癌细胞或其他致病细胞,从而达到治疗癌症或其他疾病的目的[ 18−20 ]。
CART19疗法的开发基本遵循“一个基因,一种药物,一种疾病”的理念。对于CD19 CAR构建体的创建,CD19 CAR构建体是活性药物成分(active pharmaceutical ingredient,API)(图2和图3)[ 21,22 ]。然而,这是非常规的,因为CD19 CAR需要封装在减毒慢病毒或其他类型的载体中,然后被转导到患者自己的T细胞中[ 23,24 ]。这与常规药物制剂大不相同,常规药物制剂将活性药物成分与其他惰性成分结合在一起。常规药物制剂的目标是改善该化合物的吸收、分布、代谢与排泄(absorption, distribution, metabolism, and elimination,ADME),以最大限度地提高疗效和减少不良反应(adverse effect,AE)[25]。因为CAR转基因被永久融入T细胞基因组中,所以相应的ADME是细胞输注、转运、增殖、持续和凋亡[26]。从收集患者自己的T细胞到制作最终CAR T细胞产品的过程涉及许多复杂的步骤和质量控制,这些都难以实现自动化。建立一个能够生产最终临床级CAR T细胞产品的生产中心,与识别正确的药物靶标,为临床开发建立优化CAR同样重要[27]。这对于制药行业来说是一个全新的过程。
《图2》
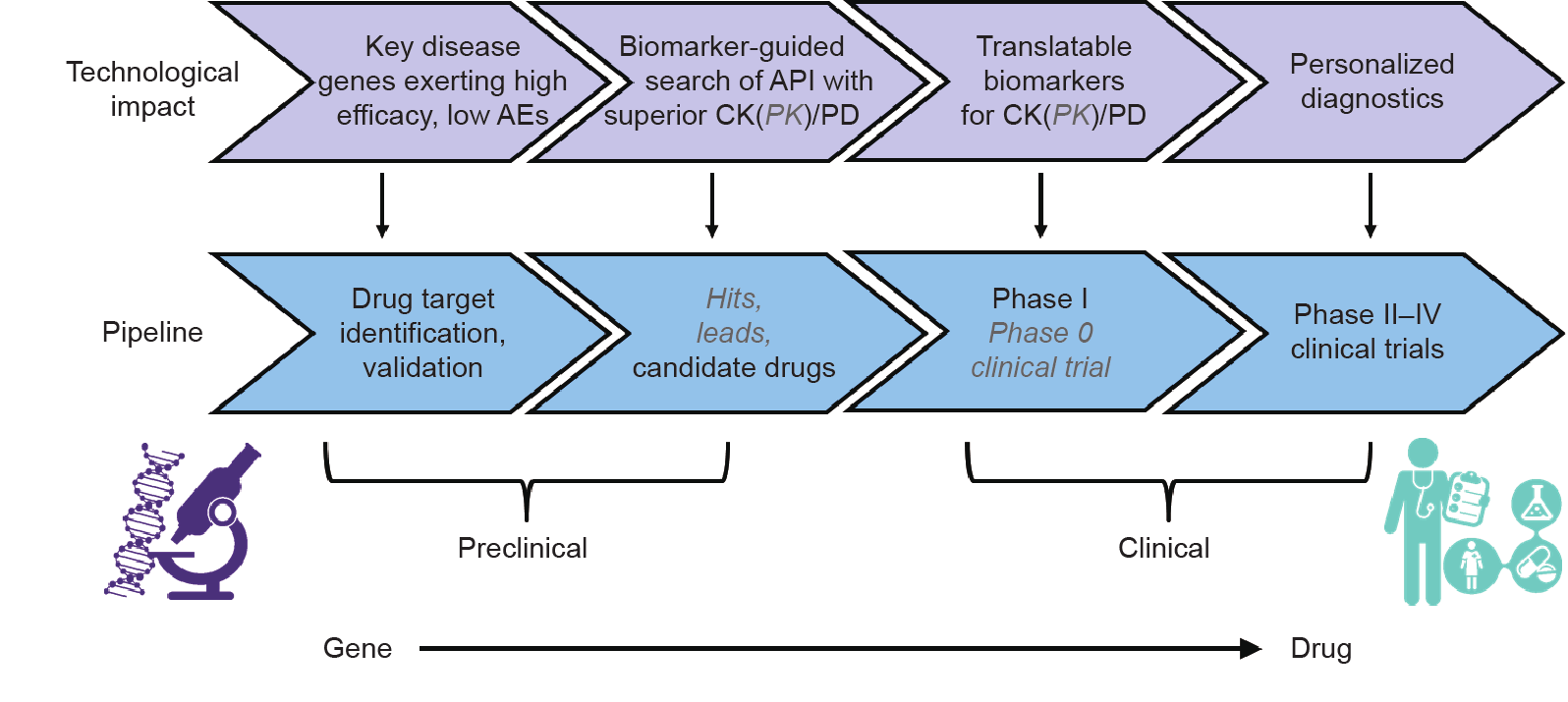
图2. 药物开发过程和技术影响,使工程化T细胞疗法开发过程与常规药物发现和开发模式相适应。目前未用于T细胞疗法的元素以灰色斜体显示。PK:药代动力学;PD:药效学;AE:不良反应;CK:细胞动力学。
《图3》
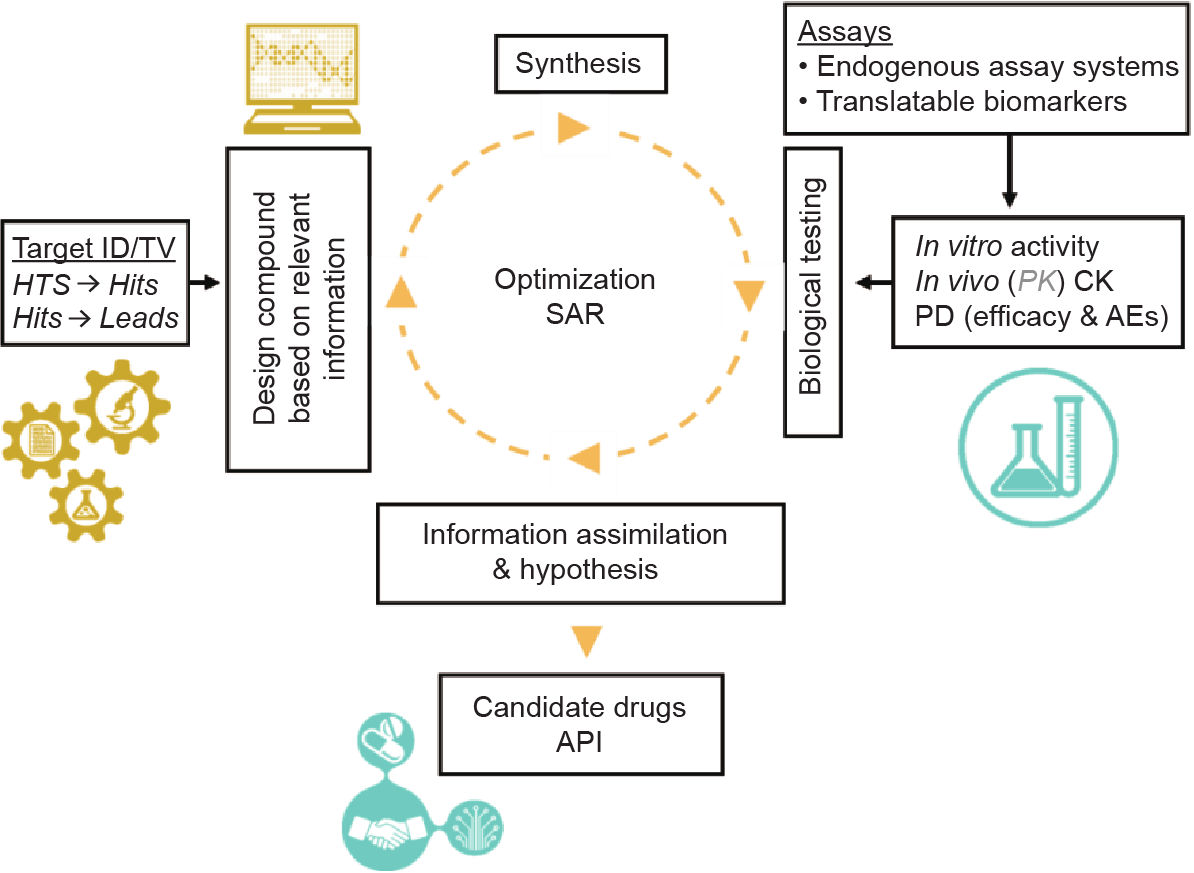
图3. 通过先导药物优化周期识别活性药物成分,将CAR构建体完善程序融入先导药物优化周期。目前未用于T细胞疗法的元素以灰色斜体显示。ID:识别;TV:靶点确证;HTS:高通量筛选;SAR:构效关系。
本文主要是以CART19为例,解释CAR的临床前和临床开发(用于工程化T细胞疗法的活性药物成分)与传统药物发现模式的相似和不同。我们还强调生物标志物在指导药物开发过程中的关键作用。此外,还讨论了先进技术和计算工具在识别更好的癌症特异性靶标、新型CAR结合结构域和生物标志物方面的影响,生物标志物可用于最大限度地提高临床疗效和降低毒性[ 28,29 ]。
《2. 工程化 T 细胞疗法的临床前开发》
2. 工程化 T 细胞疗法的临床前开发
《2.1. 靶标识别》
2.1. 靶标识别
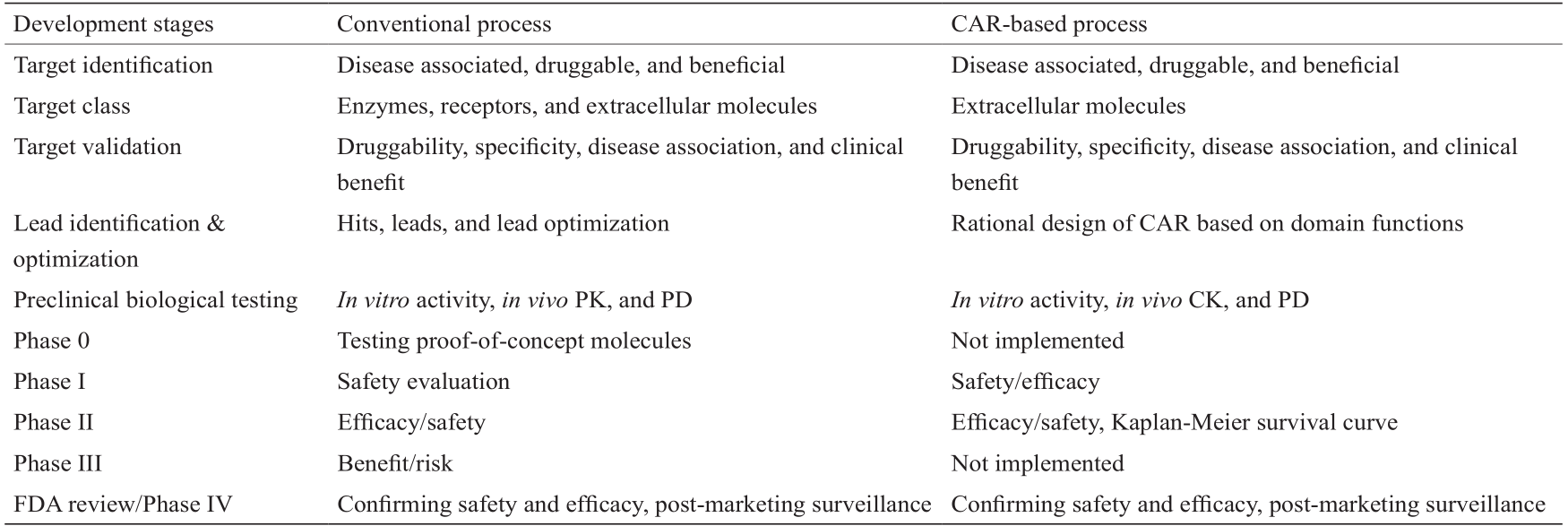
类似于常规药物开发,靶标识别是基因-药物反向药理学方法的第一步(图2、图3、表1)[ 30,31 ]。有效靶标是以最低的不良反应发生率达到最高药效的关键因素。在常规药物开发过程中,有识别理想药物靶标的一般规则。药物靶标必须与疾病相关,这意味着靶标的调节可以产生有益的临床结果。靶标必须是“可成药的”,这意味着靶标可通过外源物质调节。靶标与理想的相应物质结合将产生生物作用,从而提高临床疗效并减少临床不良反应。从传统观点来看,成药性主要与能和小分子化学物质相互作用的药物靶标有关。然而,随着科学技术的进步,单克隆抗体疗法得到了成功开发,显然,成药性真正取决于技术的可用性[ 32−34 ]。
《表1》
表1 比较传统药物与基于CAR的细胞药物的发现和开发过程
关于CAR T疗法,CAR构建体的结构域结构通常包含外域、铰链区、跨膜(transmembrane,TM)结构域和内域。外域包含抗原结合区和铰链区,而内域由共刺激区和活化区组成(图4)[ 19,35 ]。CAR构建体的结合位点/特异性通常来源于抗体对选定靶的单链片段可变(single-chain fragment variable,scFv)区。scFv可接近该区域,如CAR靶向CD19分子(CAR19)的抗人B细胞抗体(FMC63)的scFv区[ 36,37 ]。因此,只要能产生针对这些靶抗原的抗体,所有细胞外分子在CAR中均可成药。CAR理想的癌症药物靶标具有癌症特异性,位于细胞外,并与抗体相互作用。CAR经基因工程处理,可靶向细胞表面的蛋白质、糖肽和神经节苷脂[ 38−40 ]。与基于CAR的工程化T细胞疗法不同,基于工程化“转基因”TCR(tgTCR)的方法不仅限于膜抗原[41]。然而,基于CAR的疗法与抗原处理或靶细胞的主要组织相容性复合体(major histocompatibility complex,MHC)表达无关[ 35,42 ]。
《图4》
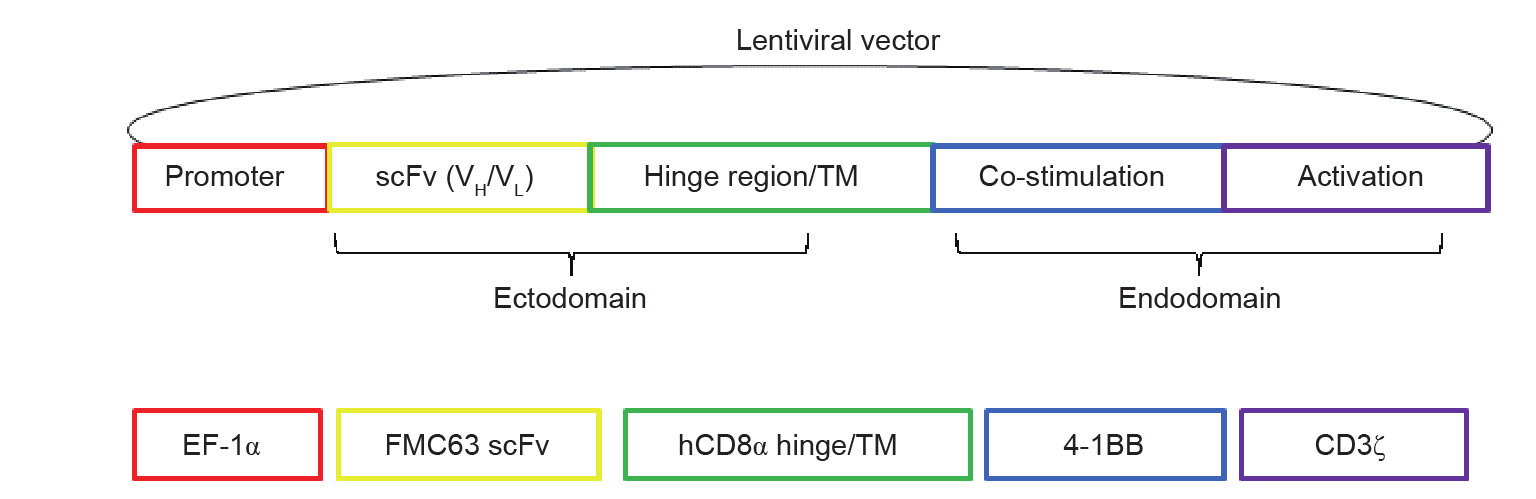
图4. 其中一个现有CAR19构建体中的主要元素。scFv:单链片段可变区;TM:跨膜;EF-1α:延伸因子-1α;hCD8α:人分化簇8α;4-1BB:人分化簇137(CD137)或肿瘤坏死因子受体超家族成员9(TNFRSF9);CD3 ζ:T细胞表面糖蛋白CD3ζ链或T细胞受体T3ζ链。
根据以下标准选择工程化T细胞疗法的癌症靶标:①新抗原,即肿瘤特异性突变,如表皮生长因子受体变体3(epidermal growth factor receptor variant 3,EG-FRvIII)[43 − 45];②在肿瘤和正常组织中表达的蛋白质,其中正常组织中缺乏蛋白靶所产生的不良反应是可控制的,如CD19 [46];③差异表达蛋白,即肿瘤中高表达的蛋白质,正常组织中较低表达的蛋白质,如人表皮生长因子受体2(human epidermal growth factor receptor 2,HER2)[47];④癌症/睾丸(cancer/testis,CT)抗原,如NY-ESO-1(一种细胞质蛋白,通过MHC I类分子加工呈递,用于TCR识别)[48,49];⑤病毒癌基因,如HPV-16 E6 [50]。
迄今为止,CAR T疗法最成功的应用是将T细胞重定向至靶向CD19。CD19是一种95 kDa单TM糖蛋白。自20世纪80年代以来,CD19被视为治疗B细胞恶性肿瘤的理想靶标[51]。氨基酸残基1~273形成CD19的胞外区,包含三个半胱氨酸二硫键和几个糖基化位点。残基274~298嵌入细胞膜中,而其余242个氨基酸位于细胞质中[52]。在细胞膜上CD21与CD81的复合物中,CD19作为B细胞受体(B cell receptor,BCR)的共受体,使BCR活性大大增强[53,54]。
CD19在除造血干细胞(hematopoietic stem cell,HSC)和某些浆细胞以外的B细胞的各个阶段均普遍表达,也在大多数B系白血病和淋巴瘤细胞中表达[51,55]。预计成功转移过继CART19将消除B系恶性肿瘤和导致B细胞再生障碍的正常B细胞。由于治疗不能根除重要的造血干细胞,并且B细胞再生障碍可通过静脉注射免疫球蛋白替代治疗,因此CD19是通过过继性转移CAR T细胞来消除B系恶性肿瘤的绝佳靶标,因为它具有抗体可及性、肿瘤特异性和非致死性正常器官组织分布[46,56]。
《2.2. 靶点确证》
2.2. 靶点确证
在过去的几十年里,基因药物开发中的传统药物靶点确证过程在制药行业已经非常系统。该过程首先评估靶标的一般信息,如靶标类别(即受体、酶等)、不同物种间的序列同源性、基因家族和组织/细胞分布。通过自然发生的突变显示药物靶标的疾病关联,某些靶标有可能出现这种情况,如硬化素[57]。药物靶标疾病的关联通过遗传模型进行证明,如基因敲除小鼠模型。在没有药物靶标的情况下,该模型将产生疗效或不良反应[30,58]。接着,在使用动物模型中的候选药物通过增加或减少药物靶标的活性来操作药物靶标时,需要证明有益的治疗结果。药物靶标在临床试验中得到进一步确证。靶点确证过程与活性药物成分的开发同步。FDA批准某种药物是对相应药物靶标的最终确证。
由于工程化T细胞疗法通过消除靶基因特异性CAR识别的靶细胞发挥其临床疗效,靶基因敲除模型与药物靶点确证的形式无关。此外,药物靶标的选择主要集中在对需要去除的细胞有特异性的分子上,如癌细胞。因此,工程化T细胞疗法的靶点确证是一个证明工程化T细胞可在细胞检测中选择性裂解预定靶细胞,并在异种移植小鼠模型中消除/抑制靶细胞生长的过程。例如,研究显示用各种CAR19构建体转导的T细胞能裂解表达CD19的细胞。通过免疫缺陷小鼠异种移植模型进一步证明了CART19细胞呈现抗B细胞恶性肿瘤[37,59]。
《2.3. 候选药物开发》
2.3. 候选药物开发
经过100多年的合成药物开发,建立了一种被称为构效关系(structure-activity relationship,SAR)先导药物优化过程的常规程序,以便选择类似药物的候选药物进行临床试验(图3)[30]。通过广泛的活体外检测筛选,在动物模型中测试具有所需生物活性和药代动力学(pharmacokinetic,PK)特性的选定化合物,以进一步确认其体内药代动力学和药效学(pharmacodynamics,PD)。药效学包括测定被测化合物的疗效和不良反应。这是一个高度依赖于现有技术和生物标志物的过程,可通过测定生物标志物显示化合物的活性。
通过合理的设计原理对这一代CAR构建体进行了设计,产生在细胞检测和小鼠模型中进行确认的先导药物候选药物(表1)[37 , 60 − 62]。评估小鼠模型和患者体内工程化T细胞的输注后药代动力学特性也很重要。然而,由于药物是活细胞形式,目前本文中采用术语细胞动力学(cellular kinetics,CK)。正如我们在最近的研究中所讨论的,最大血药浓度( )、血药浓度-时间曲线下面积(area under the plasma concentration-time curve,AUC)和最后可测血药浓度(
)、血药浓度-时间曲线下面积(area under the plasma concentration-time curve,AUC)和最后可测血药浓度(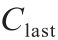 )等许多传统的药代动力学分析概念可有效应用于CK[ 26,37 ]。
)等许多传统的药代动力学分析概念可有效应用于CK[ 26,37 ]。
虽然工程化T细胞疗法仅仅处于商业化的早期阶段,但是从选择靶基因到产生候选药物的过程对于目前报道的靶标来说非常系统。在先导药物优化和候选药物选择阶段,实施的分析和技术平台与用于靶点确证的检测和技术平台相同。候选药物的开发步骤如下:首先,确定识别选定药物靶标的抗体,以提取scFv序列。其次,通过将scFv插入已建立的CAR主干构建CAR构建体。我们团队使用的一个CAR主干包含实现共刺激和活化功能的4-1BB/CD3ζ信号结构域,即CD8α铰链区和TM结构域(图4)。胞外结构域包含CD8α先导药物和药物靶标特异性scFv [ 37,59−62 ]。然后,在慢病毒包装并转导到活化人T细胞之前进行构建体的DNA序列确认,这与体外细胞扩增相结合[27]。临床前模型中广泛研究了最终CAR T细胞的活性。
Milone等[37]报道了对CAR19构建体优化的详细研究,该研究是工程化T细胞疗法候选药物/活性药物成分优化过程的教材范例。最终CAR19构建体针对其表达启动子和细胞内共刺激/活化结构域进行了优化(图4)。在体外研究中,为了选择在CD4和CD8 T细胞中提供CAR19高稳定表达的启动子,第一,他们评估了不同启动子对基因表达的影响,并选择了延伸因子-1α(EF-1α)启动子。第二,他们使用含有EF-1α启动子和各种共刺激/活化结构域的构建体,通过流式细胞术/Western和活体外细胞扩增评估了表面CD19 scFv表达。研究结果证实了由相同启动子调节的各种CAR19的相似表达水平,但在活性方面(如细胞毒性或细胞因子产生方面),并未区分含有不同共刺激/活化结构域的构建体。第三,通过细胞毒性检测评估了通过各种CAR19(CART19)转导的T细胞的细胞功能。用K562野生型(K562 wildtype,Kwt)和K562表达CD19(K19)培养CART19细胞表明,含有4-1BB/CD3ζ结构域的CART19细胞在裂解K19时效果最好,CD19阴性Kwt细胞的溶细胞活性最低。该构建体还能有效裂解表达CD19生理水平的人原发性前体B ALL细胞。第四,通过测量相应CART19的细胞因子产生,来对各种CART 19构建体进行进一步分析。与K19/Kwt一起培养表达不同CAR19的CART19 CD4+ 和CD8+ T细胞,并测量和分析细胞上清液中白细胞介素(IL)-2、干扰素(IFN)-γ、IL-4和IL-10的水平。上述研究结果清楚地表明:不同CAR19具有不同的细胞因子产生活性。第五,他们接着评估了由与K19细胞接触的CART19引发的T细胞增殖。在该检测中,共刺激/活化结构域的差异显而易见。含有4-1BB/CD3ζ结构域的CART19细胞具有CD19抗原非依赖性增殖能力。
通过人前体B ALL细胞的免疫缺陷小鼠模型对使用不同CAR19转导的CAR19进行了进一步评估。体内数据再次显示了各种CART19产品之间的功能差异,并证明了含有4-1BB/CD3ζ结构域的CART19细胞在体内具有优异的抗白血病功效。根据各种CART19产品的体外和体内数据,最终选择了含有EF-1α_4-1BB/CD3ζCAR19构建体的CART19患者特异性T细胞产品作为候选药物。
《2.4. 为 CAR19 选择 scFv 和信号结构域的历史预览》
2.4. 为 CAR19 选择 scFv 和信号结构域的历史预览
在CART19工程化时代之前,已报道了几种小鼠抗人CD19单克隆抗体(αCD19s)。Uckun等[51]广泛描述了B43单克隆抗体(monoclonal antibody,mAB),这是一种用从伯基特淋巴瘤中分离的肿瘤细胞免疫BALB/c小鼠得到的αCD19,并证明了B43识别出与B4、AB1、BU12、F974A2、HD37和SJ25-C1相同的表位。此外,克隆FMC63、HIB19和4G7是临床方法和研究中广泛使用的CD19抗体。通过用B细胞CLL(B-CLL)免疫BALB/c小鼠产生B4 mAB [63]。通过用来自毛细胞白血病患者的细胞免疫BALB/c小鼠产生HD37 mAB [55]。分别通过用NALM1 + NALM16细胞和B-CLL(BioLegend)免疫小鼠得到SJ25-C1和4G7。通过用B-CLL细胞系JVM3免疫BALB/c小鼠得到FMC63 [64]。找不到AB1、BU12、F974A2或HIB19的免疫原信息。
注意到HIB19会部分阻断B43(BD Biosciences)的结合,而FMC63已被证明会阻断HIB19或SJ25C1的结合[65]。根据现有数据必然会得出以下结论:上述所有αCD19s在类似免疫原性区域会与CD19的胞外结构域相互作用。αCD19的另一个共同之处是已知抗体的免疫原均为恶性B细胞上的人内源性CD19。
由Juno、Kite Pharama、Novartis、Fred Hutchinson癌症研究中心和中国人民解放军总医院发起的主要报道的CART19试验均采用了鼠抗CD19 scFv。在Juno发起的试验中,鼠抗CD19 scFv来自SJ25-C1,而其余试验中采用的鼠抗CD19 scFv来自FMC63 [64 , 66]。博纳吐单抗是一种FDA最近批准的双特异性T细胞衔接子,含有来源于HD37的CD19 scFv [ 55,67 ]。
在临床试验中,有患者T细胞对鼠scFv区的免疫反应报道,但没有B细胞免疫反应的报道。为了降低CAR19转基因的免疫原性,将大量研究工作集中在使scFv区人源化[ 68,69 ]。
大多数CART19试验采用了含有CD28或4-1BB共刺激结构域的第二代CAR[ 14,66 ]。为了证明4-1BB对IL-4和IL-10的诱导最小,CD19非依赖性CART19增殖,在小鼠中的植入/持久性更好以及抗肿瘤效果更好,Milone等[37]在CART19临床前评估中展示了与CD28相比的数据。在急性淋巴细胞白血病试验中,CD28和4-1BB在缓解率和毒性方面表现相似,尽管存在细微差异。利用CD28和4-1BB CART19共输注的研究可能能够描绘两个共刺激结构域的特性[70]。
《3. 工程化 T 细胞疗法的临床阶段开发》
3. 工程化 T 细胞疗法的临床阶段开发
《3.1. 工程化 T 细胞疗法临床试验设计》
3.1. 工程化 T 细胞疗法临床试验设计
对于常规药物开发,有一种测试候选药物的系统方法(图2、表1)[30]。靶向癌症疗法不同于以往的化疗/细胞毒性疗法。所述药剂的临床试验设计强调靶标结合。在试验中评估了三个水平的靶标药效学终点,包括机制验证(proof of mechanism,POM)靶标结合、原理验证(proof of principle,POP)表型变化和概念验证(proof of concept,POC)临床结果[71]。在过去的20年里,制药公司一直在努力提高成功率,减少新药的生产成本和时间。为了深入了解临床开发阶段并快速做出行与不行的决策,对不够完美的候选药物即概念验证分子(proof-of-concept molecule,POCM)进行了0期试验[72]。这门新学科已经成为众所周知的探索医学、实验医学、转化医学或发现医学。因为许多动物模型无法预测临床结果,所以该学科致力于缩小临床前动物研究与临床试验之间的差距。0期试验的主要目标是通过微剂量给药研究评估候选药物的药代动力学/药效学,对几种不够完美的候选药物进行排名,和(或)通过生物标志物确认候选药物的机制验证和原理验证[71]。
I期临床试验的主要目标是对候选药物进行安全性评估并确定其最大耐受剂量,在该临床环境中,需要记录药代动力学特性、毒性和药效学数据,I期试验通常涉及20 ~ 100名健康志愿者,研究期不到一年。II期试验的主要目标是评估药物疗效并进行进一步的安全性评估,需要进一步记录药代动力学特性、毒性和药效学数据,需纳入20 ~ 300名患者,这项研究最长持续两年。III试验的主要目标是评估关于临床结果和总体风险效益评估有效性的其他信息,需要进一步记录药代动力学特性、毒性和药效学数据,需纳入300 ~ 3000名人口统计学上不同的患者,这项研究可持续几年。IV期研究是上市后监测试验,其主要目标是持续进行安全监测和识别药物的其他用途[73]。
CART19疗法的临床试验设计是上述范例的修改版本(表1)。由于该药物是针对许多复发难治性CD19+ B细胞恶性肿瘤患者的最后手段,并且由于CART19在2017年之前是一种前景很好的实验药物,因此试验设计是上述常规试验和扩大使用的结合[74]。发表的所有I期 研 究( 如NCT01626495和NCT01029366) 均 针对绝症患者。为了证明机制验证、原理验证和概念验证,并研究CART19的细胞动力学、分化和宿主免疫原性,除了评估治疗安全性和可行性的主要目标外,I期研究还包括许多次要目标[8 , 75]。NCT01626495 II期研究NCT02435849(ELIANA)的主要目标是通过整体缓解率(overall remission rate,ORR)衡量疗效。次要目标很广泛,目的是进一步了解该疗法的疗效。正如ELIANA所证明的那样,CART19的临床效益令人印象深刻,这促使FDA批准该药物,并将其作为Kymrih出售[15]。
《3.2. 临床评估工程化 T 细胞疗法的系统性生物标志物指导程序》
3.2. 临床评估工程化 T 细胞疗法的系统性生物标志物指导程序
尽管制药业在开发CAR T细胞疗法方面还不成熟,学术研究人员已经为工程化T细胞疗法的临床开发建立了开发系统。在患者中的机制验证、原理验证和概念验证评估(进行药效学评估),CART19细胞的持久性(进行细胞动力学评估)和细胞因子释放综合征(CRS)/脱靶组织(进行不良反应/安全性评估)方面已有大量报道,本文总结如下[ 8,26,75−77 ]。
评估机制验证的方法:通过测定血清中的生物标志物评估CAR19与CDR19在靶细胞表面的结合,该生物标志物在CART19细胞与CD19+ B细胞相互作用时释放。在治疗后的不同时间点采集的患者血清和骨髓(bone marrow,BM)样品中,测定了一组包含细胞因子、趋化因子(30-plex)等因子的30种分析物[8]。IL-6、IFN-g和IL-8等分析物升高很好地显示了靶标结合。可通过商用Luminex基于微珠的检测或酶联免疫吸附检测(enzyme-linked immunosorbent assay,ELISA)测定这些分析物。此外,CART19细胞的体内扩增是另一个显示靶标结合活性的参数。采用流式细胞术通过圈选CD3+ 和CDR19+ 细胞,或通过使用CDR19构建体特异性引物对对外周血基因组DNA进行定量聚合酶链反应(quantitative polymerase chain reaction,qPCR),可测定CAR19+ 细胞的扩增[8]。
评估原理验证的方法:研究表型变化的检测包括绝对淋巴细胞计数(absolute lymphocyte count,ALC),采用流式细胞术通过圈选CD19+ 和CD20+ B细胞,或更复杂的骨髓染色和计算机断层扫描成像进行CD19+ B细胞计数。CD19+ B细胞的减少是原理验证的明显证据。
评估概念验证的方法:这些方法用于评估患者在达到机制验证和原理验证后的临床效益。关于消除肿瘤负担、无事件生存和治疗后感觉更健康的结果都是支持概念验证的证据。这并不是多余的评估,因为在实现机制验证和原理验证时,CD19 - B细胞白血病患者没有实现概念验证。
评估CAR19+ 细胞细胞动力学的方法:随着时间的推移,治疗后细胞动力学评估对于工程化T细胞疗法比药代动力学研究更为相关。采用流式细胞术在血清、骨髓或脑脊液(cerebral spinal fluid,CSF)样品的不同时间点通过圈选CD3+ 和CAR19+ 细胞可测定CAR19+ 细胞。CAR19构建体的存在也可通过qPCR或数字PCR在从外周血或其他组织提取的基因组DNA(genomic DNA,gDNA)上随时间定量,并以每微克gDNA的复制数表示。流量和qPCR数据相互关联,而PCR方法可能更加敏感。可通过上述测定推导出类似于药代动力学的参数,如AUC、半衰期(T1/2 )等[26]。
评估持续CD19特异性CAR + 细胞功能的方法:可通过使用CD107α作为标志物的脱粒检测确认持续CART19细胞的效应子功能[78]。在脱粒检测中,采集了输注后不同时间点的CART19细胞,并与靶细胞一起培养。然后使用脱粒标志物CD107α与CAR19标志物通过流式细胞术分析细胞[8]。另一种方法是测定共培养细胞上清液中的IFN-γ水平,该方法也评估了CART19细胞功能[ 37,62 ]。
评估不良反应的方法:细胞因子释放综合征是该治疗方式的主要靶标不良反应。上述30重检测或其他多重检测组合可用于监测治疗后的效果,如IL-6、IFN-γ和IL-8的升高情况。其他相关临床症状包括发烧、肌痛和恶心。某些患者由于低血压、毛细血管渗漏和缺氧,需要重症特别护理。由于铁蛋白、C反应蛋白和可溶性白细胞介素-2受体处于高水平,证明巨噬细胞活化综合征可能与细胞因子释放综合征同时发生[ 75−77 ]。
在该治疗方法中,B细胞再生障碍是一种非致死性不良反应。这是有益临床结果的标志,可通过静脉注射免疫球蛋白补充剂进行控制[46]。
CAR19的免疫原性是此类药物的一个关注点。然而,在多项研究中,输注后几个月CAR19细胞的存在表明了免疫原性的缺失[ 8,15,26,76 ]。不过,Turtle等[68]报道了一些患者对CAR19的T细胞有免疫反应。体液反应的缺失并不令人惊讶,因为治疗根除了B细胞。然而,在大多数患者中,含CAR19的小鼠scFv和来自嵌合蛋白的新交接区没有出现细胞排斥,这可能与环磷酰胺/氟达拉滨淋巴细胞清除治疗有关[79]。
神经系统不良事件在几项CART19试验中均有报道。在NCT02435849试验中,40%的患者在输注后8周内出现了一些神经系统问题[15]。在NCT02348216试验中,64%的患者有神经系统症状,其中28%为3级及以上[ 16 ]。Juno的ROCKet试验因患者脑水肿死亡而暂停[80]。Gust等[81]报道了神经毒性的风险因素,如内皮激活和血脑屏障增加。
《4. 工程化 T 细胞疗法的技术影响》
4. 工程化 T 细胞疗法的技术影响
CART19的胜利仅仅标志着工程化T细胞疗法行业的开始。我们预期,随着技术和人工智能的不断进步,该行业将不断成长和成熟。以下列举了技术和计算机辅助分析工具不断增强的一些领域。
《4.1. 通过基因组学和生物信息学工具扩展新的癌症特异性靶标》
4.1. 通过基因组学和生物信息学工具扩展新的癌症特异性靶标
在后基因组时代,先进的基因组技术和生物信息学必将对肿瘤特异性靶标的识别产生影响。研究证明,基于质谱分析的多重蛋白靶向分析方法可用于鉴定癌症特异性突变蛋白。该方法可用于鉴定开发诊断试剂盒的癌症生物标志物或靶向癌症治疗的新型药物[82]。新抗原主要通过全外显子组测序和生物信息学分析进行鉴定。此外,当该方法通过共培养多克隆TIL群体和用源于新抗原的串联微基因转染的抗原呈递细胞与TIL筛选相结合时,可以鉴定出平行的单细胞RNA序列、新抗原特异性TCR和相应的新抗原[83 , 84]。通过对多个公共转录组数据库的深度挖掘,在19种癌症类型中发现了具有肿瘤-睾丸表达模式的新基因[85]。
《4.2. 新型 CAR 结合结构域鉴定:组合抗体库的 HTS》
4.2. 新型 CAR 结合结构域鉴定:组合抗体库的 HTS
虽然目前报道的CAR(如CAR19)是根据现有抗体构建的,但高通量筛选(high-throughput screening,HTS)和先导药物优化过程有望在未来的工程化T细胞疗法开发中发挥关键作用(图3)[37 , 86]。小分子药物发现的筛选概念和技术可随时用于鉴定理想的CAR结合结构域[87]。正如组合化合物库为小分子候选药物鉴定提供了新的化学平台一样,组合全人源抗体库将成为挖掘含有新结合位点或不同结合亲和力的scFv的来源,可用于构建具有所需特异性或亲和力的CAR[88 − 90]。HTS相关技术和计算工具有望在扩大工程化T细胞疗法组合中发挥重要作用。
《4.3. 发现生物标志物以预测细胞因子释放综合征的发作并协助管理计划》
4.3. 发现生物标志物以预测细胞因子释放综合征的发作并协助管理计划
CART19治疗相关的细胞因子释放综合征可以从轻微到严重,并且发作时间不同。尽早预测治疗引起的细胞因子释放综合征的能力将改善有益的临床结果并减少不良反应。在一项回顾性分析中,通过基于Luminex微珠的30种细胞因子/趋化因子和14种可溶性细胞因子受体的多重分析,确定了几个预测性生物标志物特征。例如,使用IFN-γ、可溶性gp 130(sgp130)和IL-1受体拮抗剂(IL-1RA)的三变量回归模型在输注后的前3天就可以准确预测成人组和儿童组中哪些患者出现严重细胞因子释放综合征。使用sgp130、单核细胞趋化蛋白-1(MCP1)和嗜酸粒细胞趋化因子的决策树模型也预测了患者的细胞因子释放综合征的发病情况[77]。此外,宾夕法尼亚大学和费城儿童医院的医生为细胞因子释放综合征治疗共同制定并实施了细胞因子释放综合征分级方案和治疗计划,这可以优化疗效并最大限度地降低毒性[91]。
《4.4. 通过对单个细胞上的蛋白质和转录本进行多重同时定量来鉴定生物标志物》
4.4. 通过对单个细胞上的蛋白质和转录本进行多重同时定量来鉴定生物标志物
流式细胞术的最新进展,即质谱流式细胞技术(cytometry by time-of-flight,CyTOF)有可能极大地提高分析T细胞产品的通量和广泛性[92]。虽然关于该主题的报道很少,但10X Genomics公司推出的液滴微流控系统大规模并行单细胞测序工具有望通过同时实现单细胞上蛋白质和转录体的多重定量来加速预测性生物标志物的鉴定。最近一篇关于通过测序(CITE-seq)而获得每个细胞的转录组和细胞膜蛋白表位的多重信息从而鉴定每个细胞的论文证明,使用10X Genomics平台和生物信息学分析将是一个有效策略来辨认每个免疫细胞,以便发现更有预测性的生物标记[93]。不久后发表的一篇关于使用RNA表达和蛋白测序检测(RNA expression and protein sequencing assay,REAP-seq)来分析免疫细胞的类似论文进一步支持了这一观点[94]。此外,NanoString最近公开了一种名为3D FlowTM 分析的产品,该产品能够对分选细胞进行深入的蛋白质组学和转录组学分析。
《4.5. 发现生物标志物来预测工程化 T 细胞疗法的临床结果》
4.5. 发现生物标志物来预测工程化 T 细胞疗法的临床结果
显然,药物开发的所有层面均依赖于生物标志物/检测来指导这一过程。工程化T细胞疗法中最具挑战性的问题之一是,在生产针对患者的产品之前,需要预测哪些患者可以从这种疗法中受益,因为这种疗法并不适合所有人。CART19临床产品在41名慢性淋巴细胞白血病患者的转录组学和流式细胞蛋白质组学水平、血清蛋白质组学和机能研究方面的回顾性研究取得了重大进展。通过转录组学分析发现了这组样品的潜在预测性基因标记,如早期记忆T细胞、IL-6/信号转导和转录激活因子3(signal transducer and activator of transcription 3,STAT3)基因特征与完全缓解(complete response,CR)有关,而属于已知的终末分化、衰竭、细胞凋亡和糖酵解的信号通路则与无缓解有关。对多色流式细胞术数据的客观生物信息学分析进一步显示,输注产品中CD27+ PD-1- CD8+ CAR T细胞比例较高,这与完全缓解相关。此外,在CAR T细胞生成前,CD27+ CD45RO- CD8+ T细胞水平高与完全缓解相关。这些生物标志物可能对有效生产工程化T细胞产品的过程起到重大作用[95]。
此外,在血清生物标志物的鉴定和验证中,大量的多重免疫检测方法也得到了应用。Luminex基于微珠的液相检测是同时分析至多30种分析物时最受欢迎的平台。SomaLogic蛋白组学平台利用了与抗体作用类似的经修饰DNA适配体。据报道,多重检测通过适配体同时量化一个65 μL样本中的1100多种蛋白[ 96 ]。高密度蛋白质阵列或抗体阵列等平面阵列在新生物标志物鉴定中非常有效。OriGene生产了一种蛋白阵列,其中在一张载玻片上分布有大约17 000个重复样本。CDI HuProtTM微阵列包含从一张载玻片上提取的16 152个基因的蛋白质,占蛋白质组的81%左右。这些阵列可用于血清生物标志物的发现以及其他应用。抗体阵列是新生物标志物鉴定的另一种方法,Wilson等[97]广泛讨论了其在药物发现和开发中的应用和意义。生物信息学工具在处理高含量高通量筛选时非常必要。需要通过低通量ELISA或低密度Luminex检测来验证候选生物标志物,随后可以开发出伴随或互补诊断试剂盒[98]。
全基因组关联研究(genome-wide association study,GWAS)是研究个体遗传背景的高通量基因分型技术平台。GWAS可以结合生物信息学分析在基因组层面上检测个体的数千个单核苷酸多态性(single-nucleotide polymorphism,SNP)。一项大规模群组研究的数据可以将SNP与某些临床结果联系起来。这是另一种鉴定预测性生物标志物的策略,可以作为辅助诊断并指导为候选患者制定有效的治疗方案[ 99,100 ]。
这是另一种识别预测性生物标志物的策略,可用作伴随诊断并指导候选患者形成有效的治疗方案。
FDA最近对派姆单抗和纳武单抗的批准及其诊断检测证明了伴随诊断或互补诊断的重要性,以便最大限度提高疗效,同时最大限度减少靶向治疗的不良反应[101]。预测产品是否有效以及哪些患者能从这种治疗中获益的能力,对于使工程化T细胞成为主流疗法非常重要。
《5. 未来展望》
5. 未来展望
CART19疗法成功的意义在于它能够证明T细胞经过重组后可识别并杀死导致血液恶性肿瘤以外的疾病的细胞。其治疗效果通过遗传整合到T细胞中,成为患者系统的一部分。CART19是一种非常个性化的药物。然而,单次输注的疗效有可能持续一生。随着最近基于TCR的CAR的发展,工程化T细胞疗法在对抗癌症和某些其他疾病方面的潜力将进一步提高到最大限度,这种基于TCR的CAR结合了两种平台的优点,同时还消除了每种方法的局限性[41]。
自从1909年“魔术子弹”概念的提出和通过筛选合成化学物质发现胂凡纳明以来,Paul Ehrlich最初的系统研究法已经为现代合成药物的发现和开发制定了蓝图[102]。随着生物化学方法的引入和对蛋白质功能的了解,显然酶和受体是治疗干预的良好靶标。到20世纪80年代,当分子生物学影响到生物科学的各个领域时,Paul Ehrlich提出的“魔术子弹”概念得到了充分的实现。制药行业的药物开发侧重于“一个基因,一种药物,一种疾病”的概念,最终发现了立普妥和环丙沙星等“魔术子弹”(图2和图3)。这一概念已成为20世纪现代药物开发的基础[30]。
CART19疗法的成功将Paul Ehrlich的“魔术子弹”概念带到了现代医学制造的另一个层面,并标志着一种新的药物形式的开始,即重组“活药物”。正如胂凡纳明促进了合成制药行业的出现一样,我们预计CART19疗法将促进工程化T细胞疗法的类似发展。目前有关基因组的知识以及前所未有的技术和计算创新将进一步推进这一领域,使我们能够对抗癌症和其他疾病。
自1909年“魔术子弹”概念诞生以及通过筛选合成化学品发现arsphenamine以来,Paul Ehrlich最初的系统方法已经为现代合成药物的发现和开发奠定了蓝图。
《Acknowledgements》
Acknowledgements
The authors are grateful for support from the Center for Cellular Immunotherapies and the Abramson Cancer Center at the Perelman School of Medicine, University of Pennsylvania; the Parker Institute for Cancer Immunotherapy; and Peking University. The authors thank Regina Young for coordinating the effort in the clearance of this paper.
《Compliance with ethics guidelines》
Compliance with ethics guidelines
Carl H. June: Celldex Therapeutics: Consultancy, Membership on an entity’s Board of Directors or advisory committees; Novartis Pharmaceutical Corporation: Patents & Royalties, Research Funding; Immune Design: Membership on an entity’s Board of Directors or advisory committees; Tmunity Therapeutics: Equity Ownership, Membership on an entity’s Board of Directors or advisory committees, Patents & Royalties, Research Funding. Joseph A. Fraietta and Simon F. Lacey: Novartis Pharmaceutical Corporation: Patents & Royalties, Research Funding; Tmunity Therapeutics: Research Funding. J. Joseph Melenhorst: Novartis Pharmaceutical Corporation: Patents & Royalties, Research Funding; Incyte: Research Funding; Simcere: Consultancy; Shanghai Unicar: Consultancy. Fang Chen: Novartis Pharmaceutical Corporation: Patents & Royalties. Zhongwei Xu: BIOCELTECH: Equity Ownership, Research Funding.











 京公网安备 11010502051620号
京公网安备 11010502051620号




