

《1. 引言》
1. 引言
21世纪人类面对的诸多挑战,无论是个性化医疗还是能源的产生和储存,都有一个共同的主题:材料是解决问题的关键。从传统意义上看,材料都是偶然发现或通过经验而被发现的。硫化橡胶就是一个典型的例子:19世纪时,根据观察(化合物的随机混合)发现,与硫磺等添加剂一同加热可以提高橡胶的耐久性,硫化橡胶应运而生。随着第一原则计算方法和工具的飞速发展以及计算机性能的指数级增长,科学家和工程师们现在可以真实地模拟材料在特定应用中的性质和行为,从而避免冗长的配方、合成和测试周期。这个领域被称为计算材料科学,是化学和材料科学领域中发展最快的领域之一。然而,尽管在理论方法和建模工具方面取得了长足的进步,所有可能的化学品或材料的理论空间依旧是巨大的。例如,药理相关分子的数量估计为10 60 级[1]。因此,要找到一个策略来探索这个巨大的结构空间是不可能的。
随着实验和计算数据的增加,材料信息学(MI)领域近年来发展迅速[2]。MI的一个重要任务是通过采用数学和信息科学方法[3],使用现有的材料数据来预测新材料的性质。实现这一点的关键是构建一个描述符模型,使得该模型可以基于一组已知的输入材料某些特征来预测感兴趣的属性。定量结构-性质关系(QSPR)模型是一个重要的描述符模型,其输入变量是材料结构特征。在材料性质的输入和输出之间通常存在复杂的关系,这些关系很难使用传统的线性和非线性关联方法来处理。由于机器学习(ML)方法的发展[4],这些复杂的关系现在可以有效地通过建模来找到。
ML是人工智能(AI)的分支,其目的是根据历史数据和情况来训练模型。它已经开始在材料科学中发挥重要作用,因为它能够在不知道潜在的物理机制的情况下从可用数据中获得性能和趋势。反过来讲,已经建立的ML模型也可以再次用于材料的发现和设计。ML技术在材料研究中的一些成功应用的实例包括预测钢疲劳强度[5]、合金的物理和机械性质[6]、钙钛矿材料的电子带隙[7]、催化活性[8]和酸解离常数[9],也包括有前景的多孔材料[10]、聚合物电介质[11]、混合氧化物催化剂[12]、有机发光二极管(OLED)材料[13]、超导体[14]和光伏材料[15]的鉴定。
图1 [16]描述的文献检索表明,随着在材料研究应用的增多,ML也得到了快速的发展。
《图1》
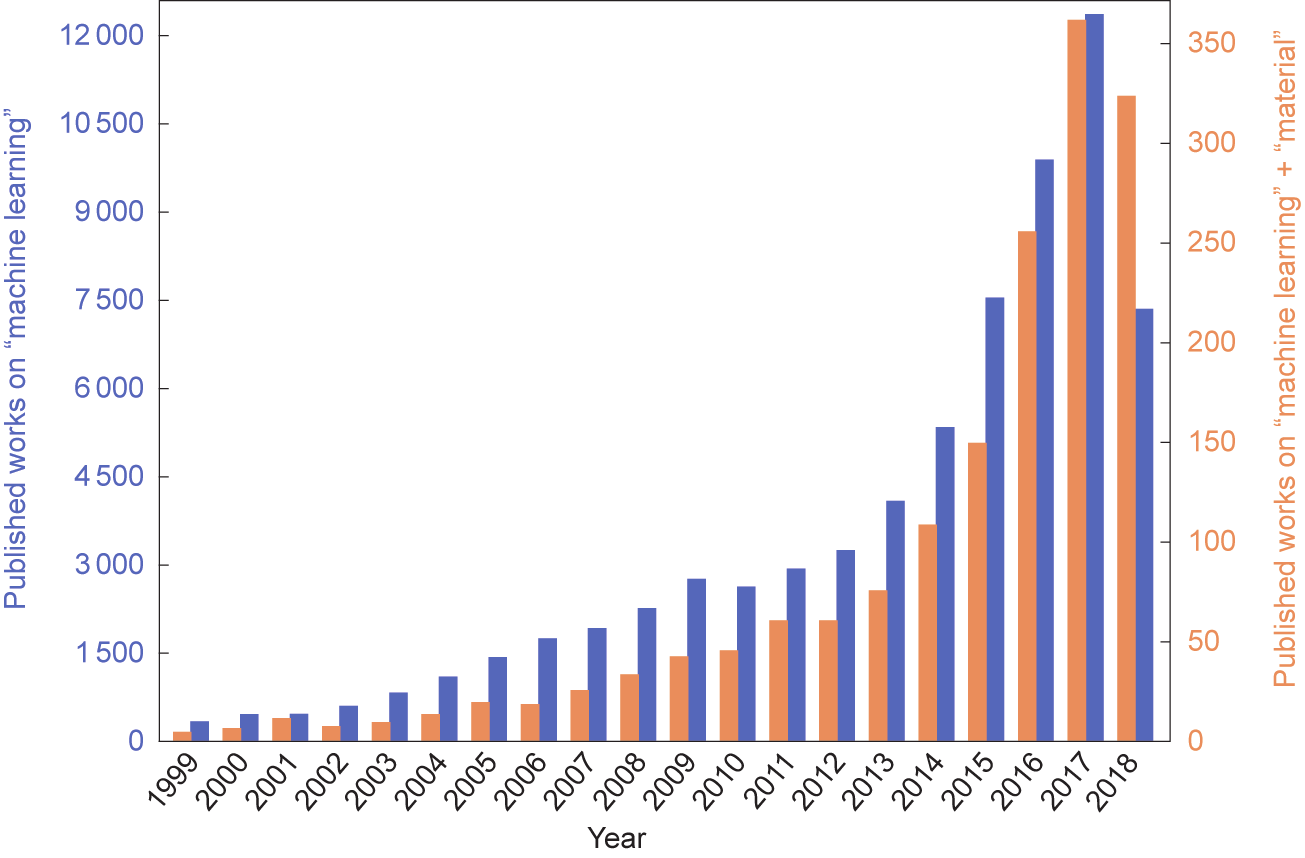
图1. 1999年1月至2018年9月出版的关于“机器学习”和“机器学习”+“材料”的著作数量。
鉴于数据驱动或ML方法在材料研究中的重要性日益增加,本文的目的是强调使用ML方法进行材料研究的主要思想和基本步骤,并概述近来ML在材料发现和设计中的重要应用。
《2. 材料科学中的大数据》
2. 材料科学中的大数据
如图2 [17]所示,几千年前,科学都是由对自然现象的经验观测组成的。几个世纪前,理论科学的范式出现了,形成了各种经典的定律、理论和模型。几十年前,随着计算机的发明,第三种科学范式即计算科学出现了,它允许根据第二种范式中总结的理论来模拟复杂的现实世界问题。材料科学的典型例子是密度泛函理论(DFT)和分子动力学(MD)模拟。在过去的几年里,大量的实验和模拟产生了第四种科学范式:连同人工智能方法一起推广的(大)数据驱动科学。机器学习作为人工智能最重要的子领域,最近几年发展十分迅速。
《图2》
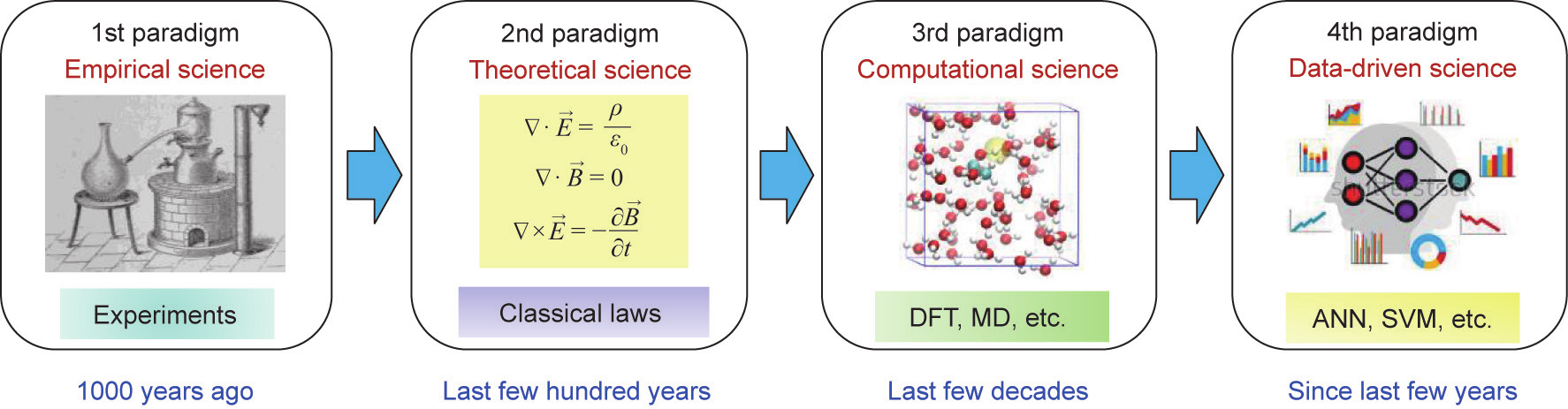
图2. 科学的四种范式:实证、理论、计算和数据驱动。ANN:人工神经网络;SVM:支持向量机。
如图3 [16]所示,“大数据”和“数据驱动”方面发表的著作数量呈现出绝对激增的状态。
《图3》
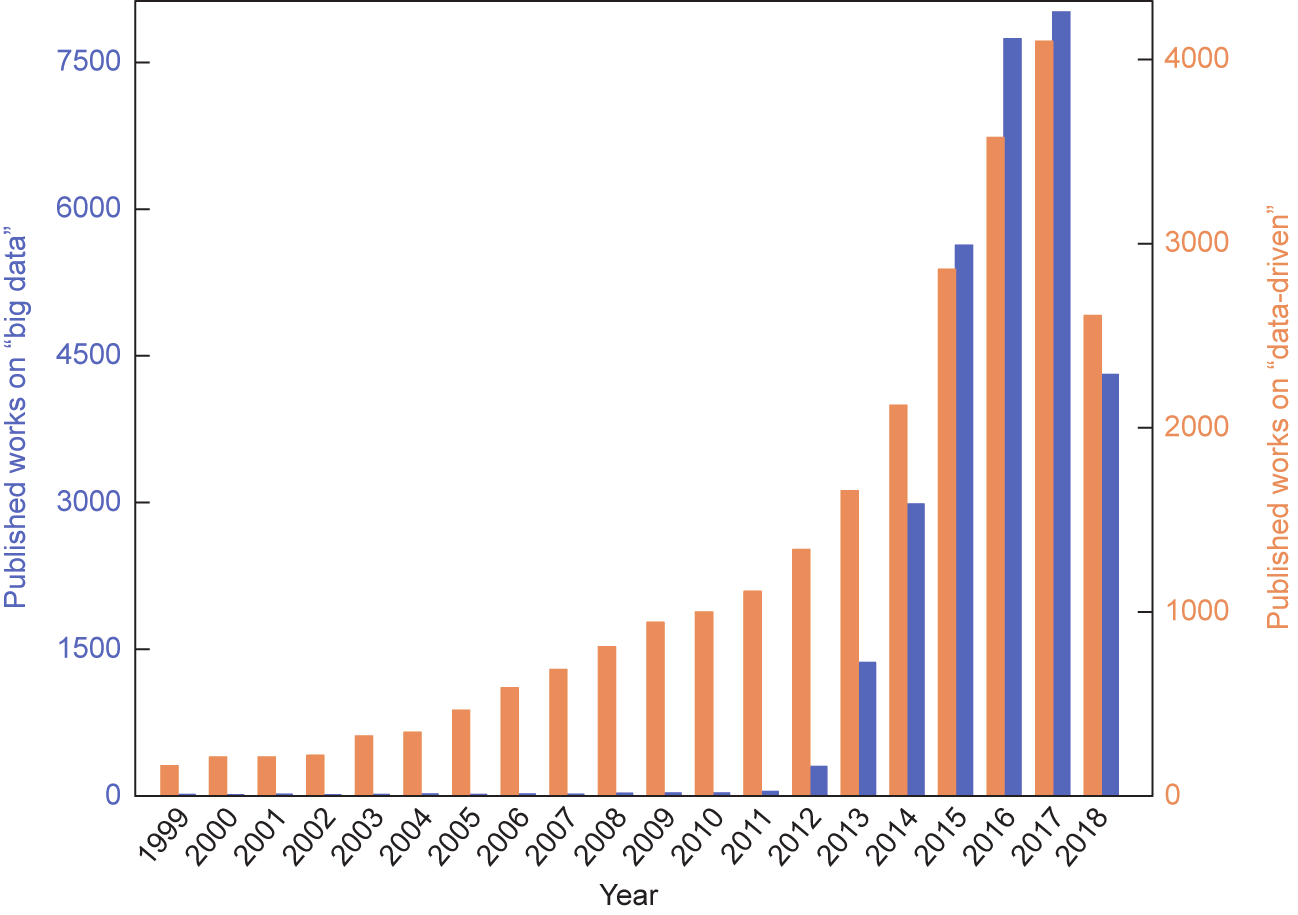
图3. “大数据”和“数据驱动”方法的出版作品数量(从1999年1月至2018年9月)。
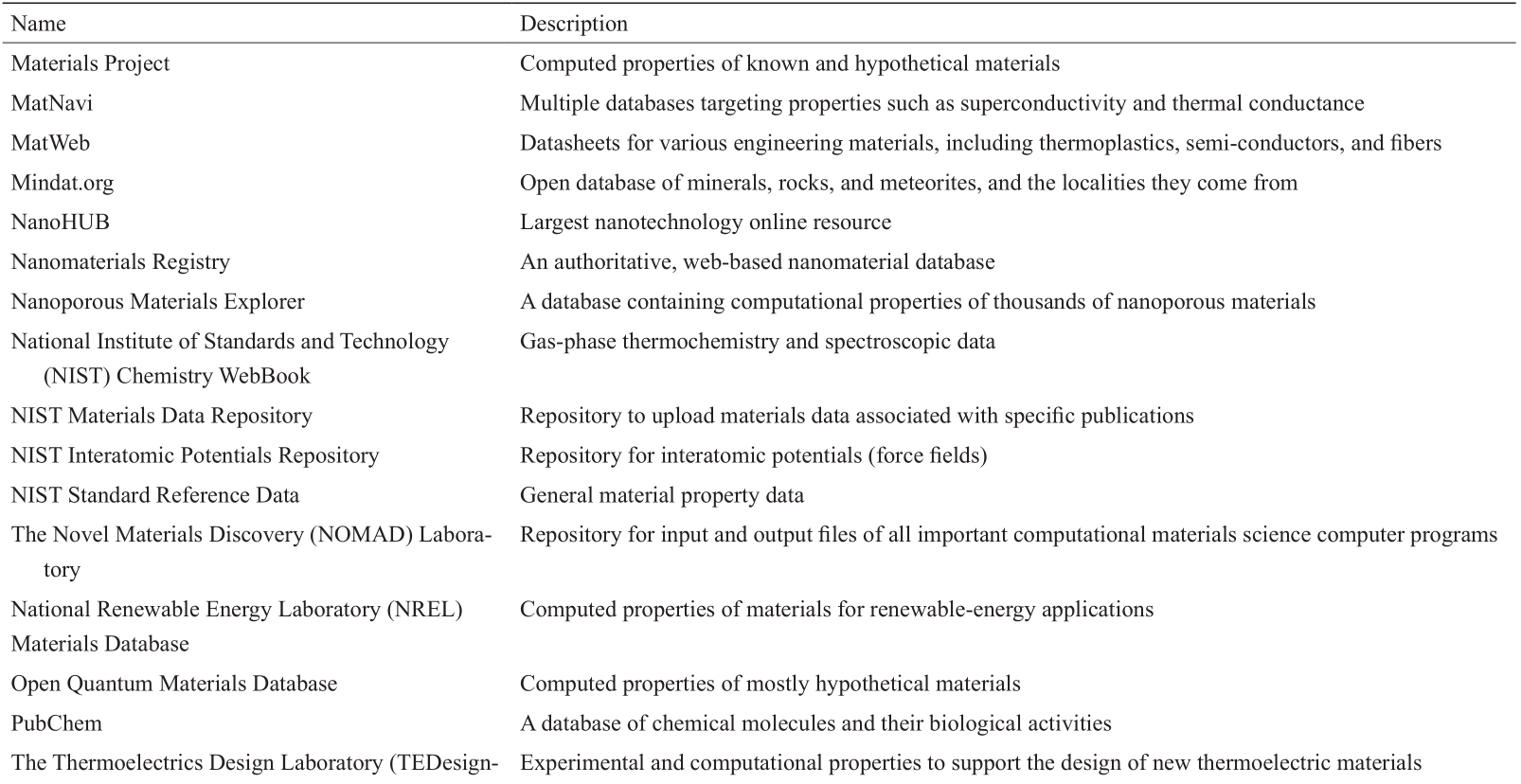
近来,材料基因组计划(MGI)和世界各地其他类似的组织活动一直在促进材料科学中大数据的可用性和可访问性。许多不同类型的材料性质数据(如物理、化学、机械、电子、热力学和结构性质)都可以由第一原则计算(如弹性模量)或实验测量(如导热系数)生成。如此大的数据为数据驱动技术或ML方法的应用提供了巨大的机会,从而加速新的先进材料的发现和设计。文献[18]中更新的表1列出了许多包含大量材料结构和特性的公开可用数据库。
《表1》
表1 分子和固体材料的公众可访问的结构和性质数据库
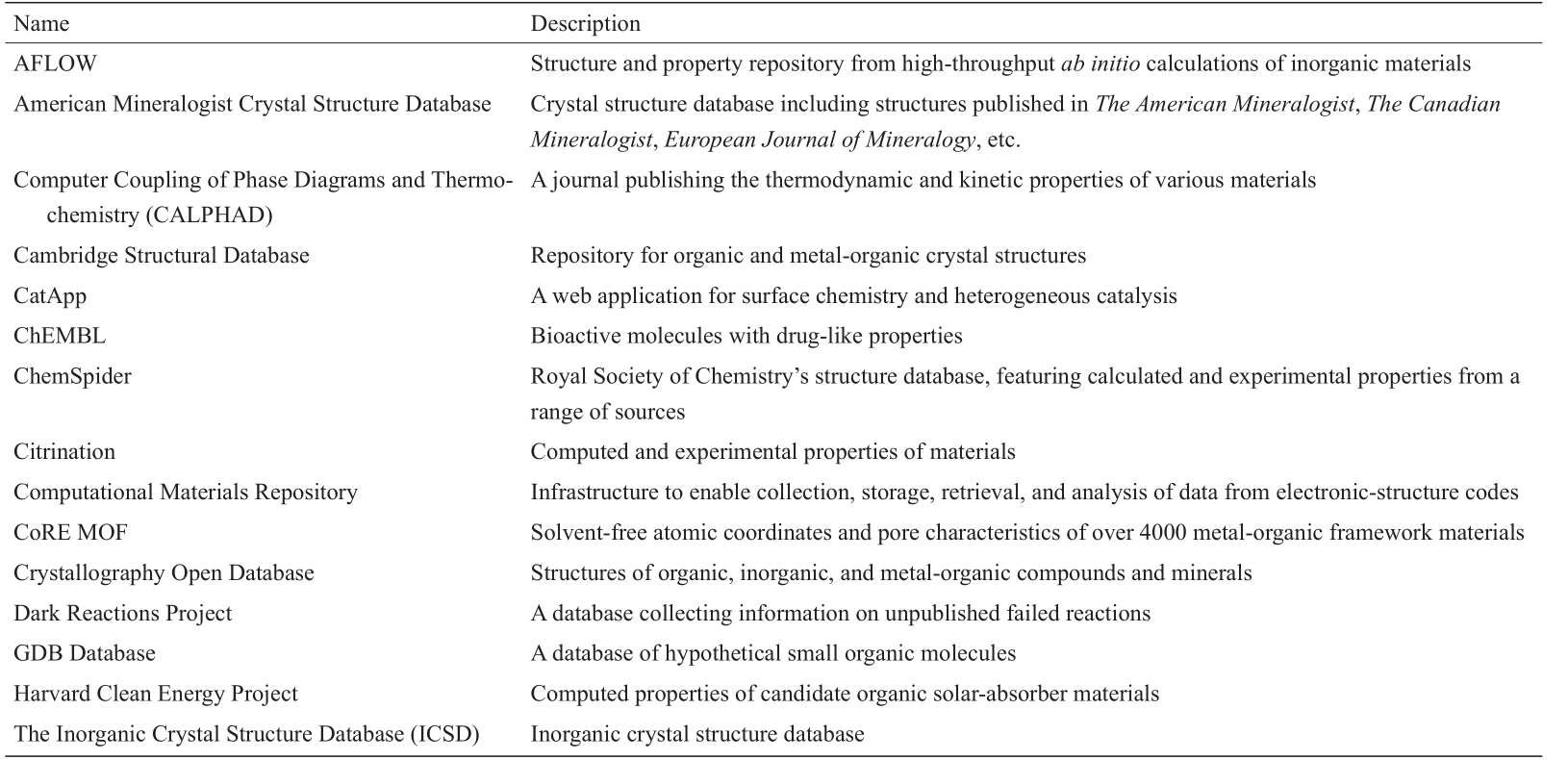
(续表)

《3. 用于材料发现和设计的机器学习》
3. 用于材料发现和设计的机器学习
现代的理论和计算工具可以有效解决众多正向问题,即在特定条件下对特定材料的特性或行为进行预测。而处理反向问题的方法和工具(如设计或制造具有特殊理想特性的新材料)则还不够完善。最近,计算机辅助分子设计(CAMD)方法[19,20]已经被提出并得到了显著发展,其目的是合理地选择或设计具有指定特性的分子。CAMD方法自从出现以来,已被用于设计溶剂,药品和消费品,工作流体,聚合物,制冷剂和过渡金属催化剂[21–31]。与CAMD问题类似,典型的材料设计任务可以如下定义:给定一个从实验和(或)第一原则计算获得的{材料→性质}数据集,具有最佳特性的最佳材料结构和成分是什么?
对于材料设计,最关键的步骤是建立一个关联模型,该模型可以基于给定的{材料→性质}数据集,准确描述输入的特定于材料的特征(通常为结构特征)与感兴趣的特性之间的关系。经典模型的构建在很大程度上依赖于物理观点和机制,例如,使用守恒定律和热力学来从现有参考数据中导出参数(通常为线性或略非线性)的数学公式。ML则采取了不同的途径:不再依赖原理或物理知识,而是仅根据现有的可用数据,就能以灵活且通常高度非线性的形式训练模型。在材料科学中,材料的结构与感兴趣的性质之间通常存在复杂的关系,且使用传统的关联方法很难处理这些关系。因此,ML方法已经成为预测材料性能、材料筛选和优化设计的重要工具。
图4展示了基于机器学习的材料发现和设计的一般工作流程。包括三个主要步骤:描述符生成和降维、模型构建和验证、材料预测和实验验证。第一步是用一组描述符或特征在数据集中表示材料。此步骤需要有关材料和应用程序的特定领域知识。第二步是在一组参考材料的已知数据的基础上,在描述符和目标属性之间建立映射模型。从简单的线性和非线性回归到高度复杂的核岭回归和神经网络,各种ML方法都可以用来建立这种映射。在最后一步中,根据所建立的ML模型进行反向设计,以找到具有期望性质的新材料。然后可以合成最佳的候选材料,并对它们的真实特性或性能进行实验验证。
《图4》

图4. 基于机器学习的材料发现和设计的通用工作流程。
《3.1. 描述符生成和降维》
3.1. 描述符生成和降维
通常,每种材料的性质都取决于一组特定要素,如晶体结构和键强度。因此,在应用ML过程之前,识别与所关注的材料特性密切相关的关键特征或描述符始终是至关重要的步骤。好的材料描述符至少应满足以下三个条件:①材料的唯一表征;②对目标特性敏感;③容易获得。根据所研究的问题或性质,可以在不同的复杂度级别上定义描述符[32]。以分子设计为例,如果正在研究非极性有机化合物的沸点或挥发性,则可以用总体水平(如总分子量)来定义描述符。如果目标是预测介电常数,则描述符可能必须包含原子级或至少基团级的信息。如果研究的是催化活性,则描述符必须包含电子级的详细信息。
Curtarolo等[33]总结了几个先前已经被开发的重要材料描述符。最简单的描述符是一维(1D)参数,如分子体积、重量和表面积、电子数量和极性。这些描述符很少或根本不包含关于材料或分子实际结构的信息。如前文所述,在预测某些属性时,更可取的是使用表示二维(2D)甚至三维(3D)结构的描述符。拓扑描述符考虑分子或材料的二维图形结构,从而反映对称性、分支和原子连通性等特征[34,35]。最常用的拓扑描述符是邻接矩阵[36]和分子连接性指数[37]。这些描述符的局限性是它们不包含任何立体化学信息。一个重要的3D材质描述符是径向分布函数(RDF)。RDF通常由g(r)表示,它定义了在另一个标记的粒子或原子[38]的r 距离处找到粒子或原子的概率。这种类型的描述符可以从实验测量(如X射线测量)和从头计算中获得。
大量数据库(表1)包含大量材料结构和属性数据。然而,应该指出的是,可用的材料数据往往彼此高度相关。因此,许多情况下,在构建ML模型之前,都有必要使用尺寸缩减工具预处理高维数据集。有几种算法[39]可为ML模型减小特征空间的维度,并帮助识别最相关的描述符(或关键特征),如主成分分析(PCA)、多维缩放(MDS)和线性判别(LDA)。例如,PCA使用正交变换将一组相关变量转换为一组简化的不相关的新变量或主成分(PC)[40]。选择每个PC时,应尽量保证它与其他PC不相关。PC构成了一个可以代表原始数据的缩小的维度空间,信息损失极小。Zhou等[41]采用PCA将12维溶剂描述符空间缩小为4维空间。于是这4 个新的描述符被成功地用于关联和预测溶剂对反应速率的影响。
《3.2. 模型构建和验证》
3.2. 模型构建和验证
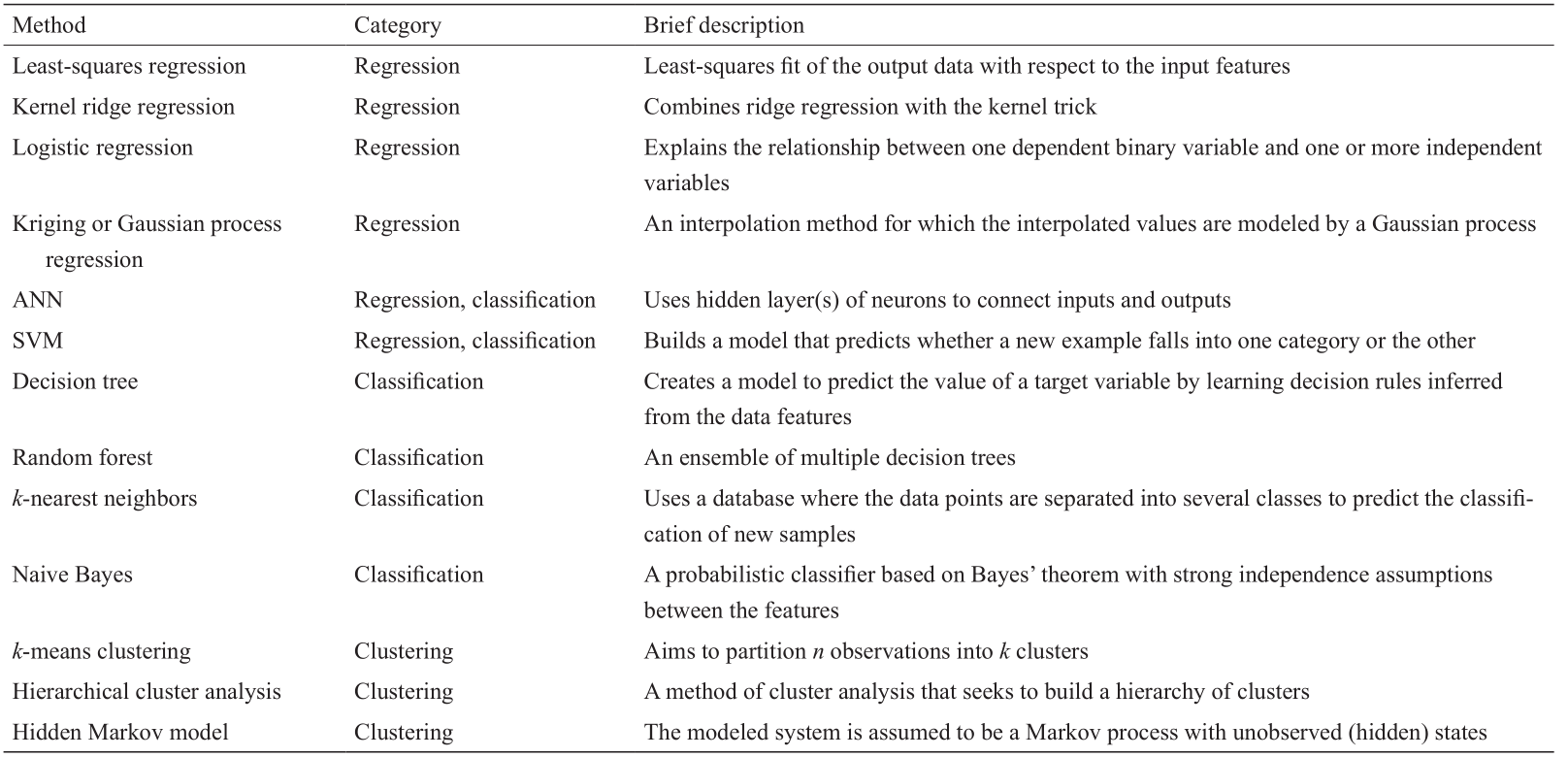
ML算法可大致分为两类:监督学习算法和无监督学习算法。监督学习可以进一步分为回归和分类。在材料设计中,监督学习试图识别一个函数,此函数能够根据一组已知材料及其属性来预测新材料属性。如果目标属性是连续量(如玻璃过渡温度),则过程称为回归。典型的回归算法有克里金或高斯过程回归[42]、人工神经网络(ANN)[43]以及支持向量机(SVM)[44]。如果输出是离散目标(如是否有毒、何种晶体),则搜索预测函数的过程称为分类。决策树[45]和随机森林[46]算法是两种最常用的分类算法。
监督学习旨在找到将一组输入数据映射到相应输出属性的函数,而无监督学习则尝试识别输入数据本身之间的关系。聚类作为典型的无监督学习方法,是将数据集划分为不同类别或区域的过程,以使同一组或群集中的数据点之间相较于与其他群集中的数据点更相似。聚类对于两个方面非常实用:一是从数据中提取物理信息;二是根据比较研究寻找有前途的新材料[47]。最流行的聚类算法是k 均值[48]和分级聚类[49],以及隐藏的马尔可夫建模[50]。
表2总结了重要的ML方法列表,且在参考文献[51]中有对每种方法的详细介绍。由于每种方法或算法都有其自身的适用性和适用范围,因此选择合适的ML算法对于其成功实施至关重要。最小二乘回归、核岭回归、神经网络和决策树这几种算法都可以创建属性预测模型。但是,某些算法(主要是基于回归的算法)提供了实际的预测功能,而其他算法(如决策树)则没有。此外,可用数据的数量也可以决定学习算法的选择。例如,要正确处理数十至数千个数据点,可以使用诸如克里金和核岭回归的回归方法。但是,当数据集比这要大得多时,则需应用更复杂的学习方法,如深度神经网络[32]。
《表2》
表2 重要ML方法的列表
近年来,许多开源软件程序或工具(如Scikit-Learn、TensorFlow和Chainer)已经被开发出来,使得非专家也可以在自己的研究中实现ML方法。Scikit-Learn是一个Python软件包,它集成了各种最先进的ML算法,包括受监督和无监督算法。TensorFlow是一个用于高性能数值计算的软件库。TensorFlow最初由Google人工智能部门的研究人员和工程师开发,如今它为ML和深度学习提供了强大的支持。Chainer是构建神经网络的有力工具,其旨在弥合算法和实现之间的差距。商业软件MATLAB的工具箱中也包含了许多ML算法,如统计学。
数据驱动模型原则上可以记住训练集中的每个数据点,从而对这些数据产生极高的精度。因此,ML模型必须利用尚未用于训练的数据。最简单的方法是进行交叉验证,即模型仅基于部分数据构建,而其余数据用于评估或验证。存在数种交叉验证策略,其中,k 折交叉验证方法[52]应用较广。在此策略中,数据集被随机划分为具有相同大小的k 个子组;k–1个子样本用于训练,其余一个子样本用于验证。此交叉验证过程重复k 次,每个子样本只使用一次作为验证数据。Kohavi [53]证明,对于实际数据集,即使计算能力允许使用更多折叠,模型验证的最佳方法仍是十倍交叉验证。另一个被广泛使用的验证ML模型的方法是自举法[54]。这种方法通过从原始数据集中逐个提取样本,并在选择样本后将其返回到数据集,构造了与原始数据集大小相同的“自举训练集”。因此,某些数据点可能会在自举训练集中多次出现,而其他数据点可能根本不显示。未在训练集中使用的数据点之后便被用于模型验证。上述过程可以重复多次,且可使用平均预测误差作为模型性能的指标。自举法的一个优点是可以用置信区间或不确定性表示结果,而其他验证方法不易使用此功能。
《3.3. 材料预测和实验验证》
3.3. 材料预测和实验验证
如图4所示,在建立ML模型后,可以执行反向设计以根据模型查找具有所需特性的材料。这可以通过使用大规模筛选或数学优化来完成。
大规模筛选方法的基本思想是:首先在设计空间中生成所有可能的候选材料,然后使用已学习的模型逐一进行测试[15]。通常,材料的生成必须考虑对材料的几个限制,这些限制通常以两种形式存在:一是结构;二是组成成分。因此,需要使用一个系统的程序来识别设计空间中的所有材料(或尽可能多的材料)。生成候选材料后,使用经过训练的模型即可简单、直接地评估其属性。
又或者可以将反向材料设计公式转化为数学优化问题,其中目标特性在受到结构和成分约束的情况下得到优化[55,56]。基于优化的方法试图在不测试设计空间中所有候选对象的情况下确定有前途的材料。而此特征使该方法受组合复杂度的限制要小得多。确定性[57]或随机算法[58]均可用于解决所确定的最优材料的成分优化问题。
确定最佳材料后,就能够合成它们并通过实验验证其实际性能。如果实验结果与预测结果吻合良好,则可以证明该材料具有最高的性能。否则将所设计的材料及相应的实验结果添加到训练集中,并重新训练ML模型。
《4. 应用实例》
4. 应用实例
ML加速了几种不同材料的开发。本文专注于三类材料:多孔聚合材料、催化材料和含能材料。以下各节重点介绍了ML方法在这些材料的发现和优化设计方面的最新应用。
《4.1. 多孔聚合材料》
4.1. 多孔聚合材料
聚合物材料具有许多合乎要求的性质,如高强度重量比、耐腐蚀性、易于成型以及较低的制造成本。由于这些优势,从传统的包装和消费产品到电气化学和生物医学工程,聚合物材料在许多工程领域有越来越多的应用。基于现有的大量聚合物结构和属性数据,数据驱动法或ML法都能在聚合物的发现和设计中发挥重要的作用。
Breneman等[59]开发了一种用于球形纳米颗粒填充聚合物优化设计的材料基因组学方法,它是以对球形纳米颗粒填充聚合物的热力学性能的预测为基础的。由实验来验证设计结果。Venkatraman和Alsberg [60]提出了一个ML模型,用来快速发现具有多种理想特性(包括高折射率)的新型聚合物材料。通过DFT计算,成功地验证了所得结果。为了促进新型聚合物材料的开发,Wu等 [61] 建立了统计模型,以预测有机聚合物的介电常数、带隙、介电损耗正切和玻璃过渡温度。一组称为无限链描述符的新特征被用于表征有机聚合物,并用作预测上述特性时的ML输入。结果表明,所有获得的ML模型均能有效地预测聚合物性能。Sukumar等[62]演示了如何构建ML模型实现具有特定电子特性的聚合物的最佳设计。模型验证证实,所建立的模型能够对训练组外的聚合物进行可靠的预测。
电介质材料传统上由无机材料制成,如瓷器、云母和石英。然而,在被用作介电材料时,聚合物具有优异的耐化学性、灵活性、廉价性和特定应用的可调性等优点。Sharma等[11]提出了一种基于ML的分级方法,以加速鉴定优于标准材料的聚合物电介质。对设计得到的聚合物的电介质性质的测量结果,有力地印证了所提出的优化聚合物电介质的方法的功效。Mannodi-Kanakkithodi等[56]以第一原则计算产生的数据为基础构建了统计学习模型,可以执行聚合物介电设计。聚合物被识别为简单的数值表示,然后使用ML算法将其映射到感兴趣的属性。此外还采用遗传算法,以进化的方式来优化聚合物的组成单元,从而直接设计出具有目标特性的聚合物。通过开发聚合物基因组,Mannodi-Kanakkithodi等[63]也构建了聚合物介电设计的基本路线图,以及向其他聚合物类别和特性扩展的未来方向。
金属有机框架(MOF)作为一种重要的多孔材料,在许多应用特别是气体储存和分离方面,有着巨大的潜力。此外,MOF的结构构建块可以组合合成几乎无限数量的材料。因此,对于MOF材料的大规模筛选和优化设计,计算方法至关重要。
Fernandez等[64]报道了储存甲烷(CH4 )的MOF的第一QSPR分析。这些学者调查了在1 bar、35 bar和100 bar(1 bar = 100 kPa)时,约为1.3 × 105 个假设的MOF下,几何特征(即孔径、表面积和孔隙率)以及框架密度对模拟CH4 存储容量的影响。基于这些数据,多个ML模型被开发出来预测MOF的CH4 存储容量,包括多线性回归模型、决策树和非线性SVM。在每种情况下,使用1 × 104 个MOF来训练模型,并用大约1.2 ×105 个MOF的测试集验证模型的准确性。结果发现,对于35 bar时的CH4 存储,理想的MOF的密度应大于0.43 g·cm–3 ,孔隙率大于0.52;对于100 bar时的CH4 存储,MOF的密度应大于0.33 g·cm–3 ,孔隙率大于0.62。基于SVM模型的响应面分析,研究人员确定了可能拥有极高CH4 存储容量的新材料。为了准确预测MOF中的二氧化碳(CO2 )的吸收量,Fernandez等[65]引入了原子属性加权RDF(AP-RDF)描述符,该描述符除几何特征外,还捕获周期材料的化学特征。基于AP-RDF描述符的非线性SVM模型有效预测了0.15 bar和1 bar下的CO2 的平衡载荷。结果表明,具有更紧凑框架和原子间距离在6~9 Å范围内的MOF在两种压力下都对CO2 表现出更高的亲和力。Ohno和Mukae [66]应用高斯过程回归来关联和预测MOF的平衡CH4 载荷。根据已建立的模型,成功确定了优于训练集中所有材料的最佳MOF。
Aghaji等[10]使用决策树和SVM方法,将材料的几何描述符作为ML输入变量,预测MOF的CO2 接收能力和CO2 /CH4 分离选择性。结果表明,孔径、空隙度和表面积是设计从CH4 中分离CO2 的最佳MOF的最重要因素。Simon等[67]使用随机森林方法发现了在氙和氪的分离方面具有巨大潜力的多孔材料。他们确定了两种高性能材料:磷酸铝沸石类似物和钙基协调网络。这两种材料都经过了合成,但尚未经过氙和氪的分离测试。Fernandez和Barnard [68]开发了用于预测MOF的CO2 和氮(N2 )吸收能力的ML模型。模型研究了许多不同的ML技术,包括决策树、k 最近邻、SVM、ANN和随机森林方法。结果发现,随机森林法对二氧化碳和氮气的接收能力都做出了最准确的预测。根据已建立的模型,确定了能够高效分离CO2 /N2 的最有前途的MOF。Qiao等[69]应用决策树法,研究了在298 K和10 bar的条件下,MOF的几何描述符与MOF分离三元气体混合物(CO2 /N2 /CH4 )时膜性能之间的关系。最终确定了7种最优的MOF膜。
《4.2. 催化材料》
4.2. 催化材料
许多工业过程中都会用到催化剂。传统地讲,催化剂的最佳设计都是凭经验或主要取决于实验。量子化学计算为第一原理催化剂设计提供了可能性。然而,巨大的计算成本将它们的应用限制在相对简单的反应和少量的候选催化剂上。随着可用的实验和计算数据的迅速增加以及催化信息学的发展,如今已可以使用ML模型很好地描述催化剂的结构和活性之间的关系,这对于催化剂的开发非常有帮助。
Huang等[70]是使用ML方法进行催化剂设计的首批尝试者之一,他们开发了一个ANN模型来描述催化剂组分和催化性能之间的关系。他们提出了一种混合遗传算法,并基于ANN模型将其应用于寻找最佳的多组分催化剂。该催化剂设计策略已成功应用于CH4 氧化偶联反应,从中发现了一些高性能催化剂,最佳催化剂的C2 烃收率达到27.78%,高于先前报道的催化剂的收率。Baumes等[71]采用ANN模型来预测水煤气变换反应的催化剂性能。结果表明,与传统的计算性和实验性反复试验方法相比,ML方法在加速发现高性能多相催化剂方面具有巨大的潜力。Baumes等[72]引入线性SVM模型来优化烯烃环氧化催化剂。之后又训练了非线性SVM模型进行第二个催化反应即轻质石蜡异构化。基于这两个应用实例,研究人员讨论了SVM与其他ML技术(如神经网络和决策树)相比在催化剂研究中的优势。
Thornton等[73]开发了一种ML模型来计算筛选3×105 种以上的用于还原CO2 的沸石催化剂。从中发现,为了使吸附时的熵焓变化最大化,最大空隙空间需大于30%以促进产物形成,同时需要大约6 Å的最佳空腔尺寸。Corma等[74]描述了如何将光谱表征描述符与常规的结构和成分描述符结合使用来构建催化剂性能预测模型。首先使用PCA从催化剂的X射线衍射(XRD)表征中提取所需的光谱描述符。然后通过使用ANN和决策树建模技术获得性能预测模型。通过将其应用于基于介孔钛(Ti)-硅酸盐催化剂的环氧化反应,证明了使用光谱描述符可以显著提高ML模型的预测准确性,从而提高催化剂设计结果的可靠性。混合金属氧化物是坚固的材料,通常用作工业催化剂。然而,先验地预测它们的催化性能是困难的。Madaan等[12]使用丁烷氧化脱氢制1,3-丁二烯作为模型反应,在实验中合成并测试了15种负载在氧化铝上的混合双金属氧化物。基于实验结果,他们建立了描述符模型,并将其用于预测1711种混合金属氧化物催化剂的性能。鉴定并通过实验验证了6种新的有前途的双金属氧化物催化剂。
双金属和多金属催化剂对各种热和电化学反应均显示出高活性。但是,对许多不同的活性部位进行建模是一个巨大的挑战。Li等[75]使用易于获得的催化剂描述符作为模型输入,开发了用于快速筛选过渡金属催化剂的ML模型。描述符包括吸附位的局部电负性和有效配位数,以及活性金属原子的固有性质,如离子电势和电子亲和力。所训练的模型被用于筛选CO2 的电化学还原所需的多金属合金,确定了几种有希望的催化剂候选。Li等[76]提出了一个以ANN为基础的框架来快速筛选双金属催化剂,模型反应是甲醇电氧化。建立了一个催化剂数据库,该数据库包含在合金表面{111}位点的*CO和*OH的吸附能以及根据DFT计算得出的活性部位的指纹特征,并将其用于优化ANN的结构和重量参数。指纹描述符包括吸附位点的sp带和d带特性,以及主体金属原子的列表特性。结果表明,使用现有的约1000 个理想合金表面数据训练的ANN模型可以捕获复杂的吸附物/金属相互作用,并在探索双金属催化剂的大型化学空间方面显示出较高的预测能力。Ulissi等[77]提出了另一个设计双金属催化剂的框架。他们列举并分类了双金属晶体每个稳定的低指数面的活性位点,从而产生了数百个可能的活性位点。同时,用基于人工神经网络的替代模型也对这些位点的活性进行了预测,发现的具有高活性的位点,为以后的DFT计算提供了目标。该设计框架被应用于电化学还原镍镓双金属化合物上的CO2 。
基于纳米材料的催化剂通常是分解成金属纳米颗粒的非均相催化剂。金属纳米粒子的表面积比其本体粒子大,因此其使用可导致催化活性增加[78]。Fernandez等[79]开发了决策树和ANN模型,根据DFT计算得出的数据集,从其结构描述符(如粒径、表面积和球形度)预测铂纳米颗粒的催化活性。结果表明,ML技术可用于快速估算纳米材料的催化性能,其分辨率是实验方法和从头算方法都无法达到的。这确定了在不久的将来指导纳米催化剂合理设计的原则或规则。众所周知,催化活性通常由一些特定的表面位点控制。因此,设计活性位点是高性能多相催化剂的实现的关键。合金纳米颗粒的活性位点分布可能不同于单晶表面上的活性位点。这使得合金纳米颗粒的最佳设计非常具有挑战性。Jinnouchi和Asahi [8]提出了一种使用局部相似性核的ML方案,这使得基于局部原子构型来了解和近似合金纳米颗粒的催化活性成为可能。该方法已成功应用于在Rh-Au合金纳米颗粒上进行的直接NO分解反应。
数据驱动的建模不仅对于非均相催化剂设计很重要,对均相催化也很重要。Maldonado和Rothenberg [80]总结了均质催化剂设计应采用预测模型的原因、时间和方式。过渡金属复合物是一种重要的均相催化剂,其具有非常复杂的电子结构,而直接对这些材料进行DFT模拟计算非常昂贵。Janet和Kulik [81]使用ANN方法来预测过渡金属复合物的电子性质,包括自旋态有序性和特定键长。结果表明,ANN的性能优于SVM和核岭回归等其他ML方法。所建立的ANN模型为大规模筛选过渡金属复合物催化剂提供了良好的基础。
《4.3. 含能材料》
4.3. 含能材料
ML在加速发现包括电池和超导体材料、电陶瓷和热电材料以及光伏和钙钛矿材料在内的高性能含能材料方面发挥着重要作用。
Fujimura等[82]基于实验和计算数据,使用ML方法来预测作为锂离子材料的锂(Li)导电氧化物的不同成分在373 K下的电导率。基于已建立的ML模型优化材料的成分,可以合理设计优质锂离子导体。晶体结构对锂离子硅酸盐阴极的物理和化学性质有很大影响,因此极大地影响了它们的电池应用。Shandiz和Gauvin [83]使用不同的分类算法预测了硅酸盐阴极的三种主要晶体类型(即单斜晶系、斜方晶系和三斜晶系)。事实证明,与其他分类方法相比,随机森林方法的预测准确性更高。Sendek等[84]提出了一种大规模的计算筛选方法,用于确定有前景的锂离子电池固态电解质的候选材料。作者首先筛选了具有高结构和化学稳定性、低电子电导率和低成本的12 831种含锂结晶固体。然后,他们使用逻辑回归建立了数据驱动的离子电导率分类模型,以进一步选择Li传导快的候选结构。候选材料的数量从12 831减少到21,其中一些已经通过实验进行了测验。Stanev等[14]使用了几种ML方案来开发不同的模型,以预测超过1.2×104 个超导体的临界温度。为了提高这些模型的准确性和可解释性,他们使用了新的描述符,这些描述符来自AFLOW在线存储库中的材料数据。最后,将回归模型和分类模型组合到一起,用于搜索整个无机晶体结构数据库(ICSD),以找到具有理想临界温度的潜在新超导体。已成功鉴定出30多种非铜酸盐和非铁基氧化物。
Scott等[85]基于最近建立的数据库,使用ANN方法设计电陶瓷材料,该数据库包含各种陶瓷化合物的组成和特性信息。随后采用随机优化算法搜索最佳材料,主要考虑高相对介电常数和低总电荷。结果发现,在某些情况下,所确定的材料与数据库中所包含的材料相似;在其他情况下,发现了全新的材料。在2.5×104 种已知材料的现有信息的基础上,Gaultois等[86]开发了一种基于ML的开源引擎,用于评估热电材料的性能。事实证明,该引擎可以识别出与已知材料不同的有前途的热电材料。
能源需求的增长以及对清洁能源的需求使太阳能电池成为重要的能源。光伏和钙钛矿材料是用于存储和利用太阳能的两种主要材料。Nagasawa等[87]使用ANN和随机森林模型筛选了用于有机光伏应用的共轭分子。他们从文献中收集了包括分子量、电子性能和功率转换效率在内的参数,并进行了机器学习。结果表明,随机森林模型比基于ANN的模型的预测精度高。Olivares-Amaya等[15]使用ML技术开发模型,用来预测潜在有机光伏分子的重要电流电压和效率特性。所得到的模型将用于从2.6×106 种候选化合物中快速筛选有前途的光伏材料。结果表明,苯并噻二唑和噻吩吡咯同系物是目前光伏应用最有前途的分子。Yosipof等[88]提出了一种数据挖掘和ML工作流程,并将其用于分析两个最近开发的基于Ti和铜氧化物的太阳能电池库。结果表明,由k 最近邻算法构建的ML模型可以有效预测多种太阳能电池特性。因此,该模型适合在新的有前途的金属氧化物的基础上,设计更好的光伏太阳能电池。
钙钛矿太阳能电池是另一种类型的太阳能电池,它包含作为光收集活性层[89]的钙钛矿结构化合物(最常见的是基于有机-无机铅或卤化锡的混合材料)。双钙钛矿带隙的准确预测对于它们的太阳能电池应用非常重要。用于量化带隙的量子力学计算非常昂贵,因而数据驱动的ML方法成为有前途的替代方法。Pilania等[7]开发了一个稳健的ML框架,用于高效、准确地预测双钙钛矿的电子带隙。经过验证,已建立的学习模型可用于设计有前途的用于太阳能电池的钙钛矿材料。居里温度( )是二阶相变温度,是钙钛矿材料的另一个重要物理特性。Zhai等[55]采用SVM、相关向量机和随机森林方法来建立 的预测模型。根据k 折交叉验证,SVM模型显示出比其他两个模型更好的预测性能。通过使用遗传算法来指导搜索,发现了具有高 的潜在钙钛矿材料。
)是二阶相变温度,是钙钛矿材料的另一个重要物理特性。Zhai等[55]采用SVM、相关向量机和随机森林方法来建立 的预测模型。根据k 折交叉验证,SVM模型显示出比其他两个模型更好的预测性能。通过使用遗传算法来指导搜索,发现了具有高 的潜在钙钛矿材料。
《5. 结论》
5. 结论
数据驱动科学是科学的第四范式,它产生了MGI和材料信息学。MGI和材料信息学的发展已经完全改变了材料研究和发展的理念。现在,科研人员不再依赖实验性的反复试验或高通量的从头计算,而是利用数据驱动或ML方法预测各种材料的性质并指导实验人员发现和开发新的高性能材料。本文简要介绍了不同类别的ML算法以及相关的软件和工具。总结了利用ML方法进行材料发现和设计的基本步骤。重点介绍了多孔聚合材料、催化材料和含能材料的大规模筛选和合理设计的最新应用。尽管已拥有大量成功的应用,这个课题仍处于起步阶段;在可预见的未来中,相信ML会在加速各种功能材料的开发中扮演越来越重要的角色。
《Acknowledgement》
Acknowledgement
The authors acknowledge the financial support from Max Planck Society, Germany.
《Compliance with ethics guidelines》
Compliance with ethics guidelines
Teng Zhou, Zhen Song, and Kai Sundmacher declare that they have no conflict of interest or financial conflicts to
disclose.











 京公网安备 11010502051620号
京公网安备 11010502051620号




