

《1、 引言》
1、 引言
全球每年产生数千亿吨的废弃木质纤维素生物质,但其利用率不足10%。巨大的生产量和燃烧处理给环境带来了严重的污染,因此,废弃生物质的转化得到了全球范围内的关注[1‒2]。当前,除了将生物质糖化生产乙醇外,许多类型的生物质被转化为能够用于环境修复的碳材料[3]。生物质在限氧条件下高温热解可以生成具有高比表面积、丰富孔隙和富官能团的生物炭(biochar, BC),它们被当作环境功能材料广泛应用到废水处理中[4]。为了提高BC对污染物的降解效率,负载纳米零价铁(nZVI)的生物炭材料应运而生。nZVI-BC这类复合材料在染料、重金属和有机污染物的降解中表现出优异的去除性能[5‒7]。
值得注意的是,许多研究表明nZVI-BC材料的制备存在某些缺点,热解过程中不完全还原或获得材料后的易氧化会造成nZVI-BC应用效能的降低[8‒9]。nZVI-BC的常见制备方法大致分为两种类型:液相还原和碳热还原。这两种方式的区别在于铁盐改性的对象的不同,液相还原针对的是生物炭的改性,碳热还原针对的是生物质的改性。液相还原的步骤为:首先热解生物质为生物炭,然后在N2气氛下,借助硼氢化物(NaBH4或KBH4)将Fe3+或Fe2+盐还原为Fe0并负载至生物炭上,最终制备出nZVI-BC [10]。该方法的缺点是过程烦琐且Fe0易被氧化,需要在真空下储存[11‒12]。相比之下,碳热还原法是先采用浸渍法改性生物质,然后将改性的生物质热解为nZVI-BC [13]。该方法虽然减少了硼氢化物和N2的利用,但它仍有一些缺点,比如由于生物质的顽固结构造成改性过程中铁盐的使用量大和浸渍反应时间长的问题。
生物质由三种主要成分组成,分别是纤维素、半纤维素和木质素。它们通过强烈的共价键和氢键交织在一起,导致了生物质的异质性[14‒15]。此外,木质素包裹在生物质的外层,赋予生物质表面坚硬和光滑的特性,降低了其他物质的可及性[16]。因此,实现碳热还原的前提是通过对生物质预处理提高其表面可及性,获得含有氧化性铁的改性生物质。研究表明,石墨化碳可以通过充当材料的“壳”来保护纳米铁颗粒免受氧化,同时金属铁作为“核”为碳骨架提供强大的支撑,防止外部坍塌[14]。生物炭的碳骨架主要来自木质素的热解,木质素由许多芳香环组成,热解温度范围为165~900 °C [17‒18]。因此,若要实现nZVI-BC的改进可以从以下两个思路入手:首先是在生物质改性过程中,通过铁盐预处理的生物质需要尽可能多地保留木质素以保证后续热解的芳环骨架;其次是在热解过程中,氧化性铁需要依靠充分的还原性物质将其还原为ZVI。然而,在碳热还原法制备nZVI-BC的研究中,对于详细的铁盐改性生物质及热解制备nZVI-BC的机制的研究十分有限。
基于许多生物质预处理策略(酸、碱和有机溶剂处理)[19]和木质素去除机制[20]及传统的生物质浸渍改性方法(铁沉积)[21],我们推测,在生物质和铁盐的非均相改性体系中引入特定的溶剂,同时减弱预处理条件(时间、温度等),可同时满足改善生物质结构保留较多的木质素以及提高氧化性铁的含量。
在该研究中,我们首次提出了一种新的RS改性策略。一种绿色溶剂PEG400以催化剂的形式被引入传统的氯化铁改性RS的体系。PEG400是一种无毒的线性聚合物,基本上不挥发[22];并且它被认为是一种相转移催化剂,能够在水性双相系统中分离和螯合金属离子[23]。基于该改性方法获得的生物质在碳热还原后转化为具有核壳结构的nZVI-BC。nZVI-BC的物理化学性质被表征。RS改性和nZVI-BC形成的机制通过研究RS的结构变化、铁相转移和原位还原被阐明。此外,在染料去除实验中测试了nZVI-BC的催化应用潜力,实验结果表明nZVI-BC具有优异的吸附和催化能力。本研究为nZVI-BC的制备提供了新的见解,在一定程度上能推动该材料的实际生产和应用,对生物质转化为环境功能碳材料并应用于环境修复具有重要意义。
《2、 材料和方法》
2、 材料和方法
《2.1 生物质和化学品》
2.1 生物质和化学品
RS购自黑龙江省五常市。RS通过粉碎机切割粉碎,过筛(40~100目)后作为原料。在使用前,RS原料被洗涤数次以除去灰分,然后干燥备用。PEG400 [分析试剂(AR),≥99.9%]和FeCl3·6H2O [AR, ≥99%]购自国药集团化学试剂有限公司。所有实验用水均为超纯水。
《2.2 RS改性》
2.2 RS改性
称取干燥的RS [5 wt% (m/V)]到圆底烧瓶中,与PEG400 [1:1 (V/V)]和FeCl3·6H2O (2.5%, m/V)的溶液混合。然后置于油浴锅中,在设置的温度和恒定磁力搅拌下进行处理。反应结束后,通过真空过滤进行固液分离。最后,使用大量水反复冲洗处理后的固体RS以去除PEG400和多余的FeCl3,直到pH值为中性。洗涤后的RS在烘箱中于60 ℃下干燥24 h。具体实施的对照实验有:①未预处理RS;②单独用FeCl3·6H2O溶液预处理RS(80 ℃, 1 h);③用FeCl3·6H2O和PEG400预处理RS(60 ℃/80 ℃/100 ℃, 0.5 h)。从上述三种预处理中获得的样品被分别命名为RS、F-RS和PFx-RS,其中,x表示预处理温度为60 ℃、80 ℃和100 °C。
《2.3 热解制备nZVI-BC》
2.3 热解制备nZVI-BC
在热解之前将预处理后的RS(RS、F-RS和PFx-RS)完全干燥。然后,将它们转移到刚玉方舟并置于管式炉中,在N2气氛条件中,以5 ℃∙min-1的速率升温至700 ℃并保持2 h。热解完成,待生物炭冷却至室温时取出。将其研磨并过0.154 mm筛网,然后储存在密闭容器中以进一步使用。获得的生物炭分别命名为BC、FBC和PFxBC,PFx-BC中的x表示RS的改性温度为60 ℃、80 ℃和100 ℃。
《2.4 生物质表征》
2.4 生物质表征
为了阐明nZVI-BC的形成机制,考察了改性前后RS的理化特征变化。能量色散X射线光谱(EDS, OXFORD X-Max,英国)用于观察RS的元素分布和含量变化,特别是铁含量的变化。X射线光电子能谱(XPS, Kratos Axis Ultra DLD,英国)被用于研究化学元素变化,并通过分峰拟合确定铁相的组成。
《2.5 生物炭表征》
2.5 生物炭表征
生物炭的形貌和内部结构特性通过配备有EDS(OXFORD X-Max,英国)的扫描电子显微镜(SEM, Zeiss Sigma 500,德国)和透射电子显微镜(TEM, JEM1400,日本)来表征。物相组成通过X射线衍射测试(XRD, Bruker D8 Advance,德国)来确定。nZVI-BC的磁场强度通过振动样品磁力计(VSM, Lake Shore 7404,美国)来监测。nZVI-BC表面上的电荷分布通过Zeta电位计(Malvern Zetasizer Nano S90,英国)来测量。表面元素含量和化合价变化通过XPS来监测。生物炭的官能团通过傅里叶变换红外光谱(FTIR, PerkinElmer Spectrum One,美国)来分析。比表面积、孔体积和孔径分布通过N2吸脱附等温线(BET, Quantachrome NOVA Station B,德国)来监测。碳材料的结构及石墨化程度通过激光共聚焦拉曼显微光谱(Raman, inVia-Reflex,英国)在室温、532 nm的激发波长下来表征。
《2.6 刚果红的去除》
2.6 刚果红的去除
nZVI-BC被作为催化剂催化过硫酸盐氧化去除刚果红(CR)。反应条件如下:CR浓度为30 mg·L-1,pH为6.8±0.2,反应时间为0~150 min,nZVI-BC的投加量为0.5 g·L-1,过二硫酸钾(K2S2O8; PS)的浓度为 1.8 mmol·L-1,反应体积为50 mL,温度为30 ℃。将nZVI-BC和PS按设定的比例加入塑料白瓶中,并将白瓶放入水浴摇床中。在设定的时间间隔内,取出500 µL溶液稀释,在8000 r·min-1下离心3 min。然后使用紫外-可见分光光度计(A360)在波长498 nm处分析其吸收强度。CR的去除率根据公式(1)计算:
(1)
式中,C0和Ct分别是溶液中CR的初始浓度和t(min)时刻的浓度。对照实验为仅有PS的氧化实验和单一nZVI-BC的吸附实验。反应前后的材料通过XRD、FTIR和SEM、EDS表征。
《3、 结果和讨论》
3、 结果和讨论
《3.1 nZVI-BC的理化性质》
3.1 nZVI-BC的理化性质
根据设计的实验过程(附录A中的图S1),首先将RS在FeCl3溶液(80 ℃,1 h)中以及在PEG400和FeCl3混合溶液(60 ℃/80 ℃/100 ℃, 0.5 h)中加热处理改性,然后改性后的RS通过一步热解法转化为相应的材料,分别命名为FBC、PF60BC、PF80BC和PF100BC。
《3.1.1. 物理特性》
3.1.1. 物理特性
(1)XRD分析。PF60BC、PF80BC和PF100BC的XRD谱图[图1(a)]在约44.7°处显示出特征峰,对应于Fe0典型的(110)面;这与以前报道的结果一致[24‒25]。随着温度的升高,峰值变得越来越强,表明生物炭中Fe0含量的增加。另外,存在于PF100BC谱图中的65.0°和82.3°处的衍射峰指向Fe0的(200)和(211)面[26]。然而FBC谱图中没有出现Fe0的衍射峰,表明单一FeCl3改性不如PEG400和FeCl3混合溶液的改性。以上结果证明了nZVI-BC的成功制备。此外,值得注意的是PFxBC的物相组成不含其他杂质,优于文献报道的一些类似材料[8‒9,12]。
《图1》

图1 所得材料的XRD(a)、VSM(b)和拉曼光谱(c)谱图。
(2)VSM分析。VSM结果[图1(b)]显示PFxBC是具备磁性的,但FBC几乎没有磁性。这一趋势与XRD结果一致。结合XRD的物相分析,可以认为PFxBC的磁性来自Fe0。PFxBC的磁化强度从PF60BC(3.00 emu·g-1)逐渐增加到PF100BC(4.94 emu·g-1),是FBC(1.08 emu·g-1)的4.57倍。虽然该材料的磁化强度不如其他一些负载氧化铁的磁性生物炭的那么高[27],但它仍然与报道的大多数磁性生物炭相当[28‒29],在实际测试中证实了该nZVI-BC可以通过磁力从液相系统中分离。
(3)拉曼分析。拉曼光谱[图1(c)]测试了四种生物炭的碳结构和石墨化程度。D峰(1360 cm-1)和G峰(1590 cm-1)分别归因于无定形碳和石墨碳[30]。ID/IG从FBC的0.696增加到PF60BC的0.746、PF80BC的0.783和PF100BC的0.799,表明Fe0的产生导致生物炭的石墨化程度降低。随着Fe0强度的增加,ID/IG不断增加,将更多的无序结构引入nZVI-BC。
(4)SEM和TEM分析。BC、FBC和PF100BC形貌的SEM图像显示在附录A中的图S2(a)和图2(a)~(c)中用于比较。BC [图S2(a)]和FBC [图2(a)]的表面光滑,几乎没有颗粒。相比之下,PF100BC [图2(c)]中嵌入了大量的白色颗粒。通过与PF80BC的比较[图S2(c)]进一步证实了这一观察结果,PF80BC绿色框内的白色颗粒不如PF100BC中的多。通过TEM [图2(d)]分析,我们证实PF100BC内部生长的大量白色颗粒是nZVI颗粒,这一结果与其他研究一致[31‒32]。此外,Fe0颗粒为纳米级,尺寸低于100 nm。PF100BC表面的白色颗粒被认为是其他铁相成分,如氧化铁;XPS证实了这种颗粒的存在。
《图2》
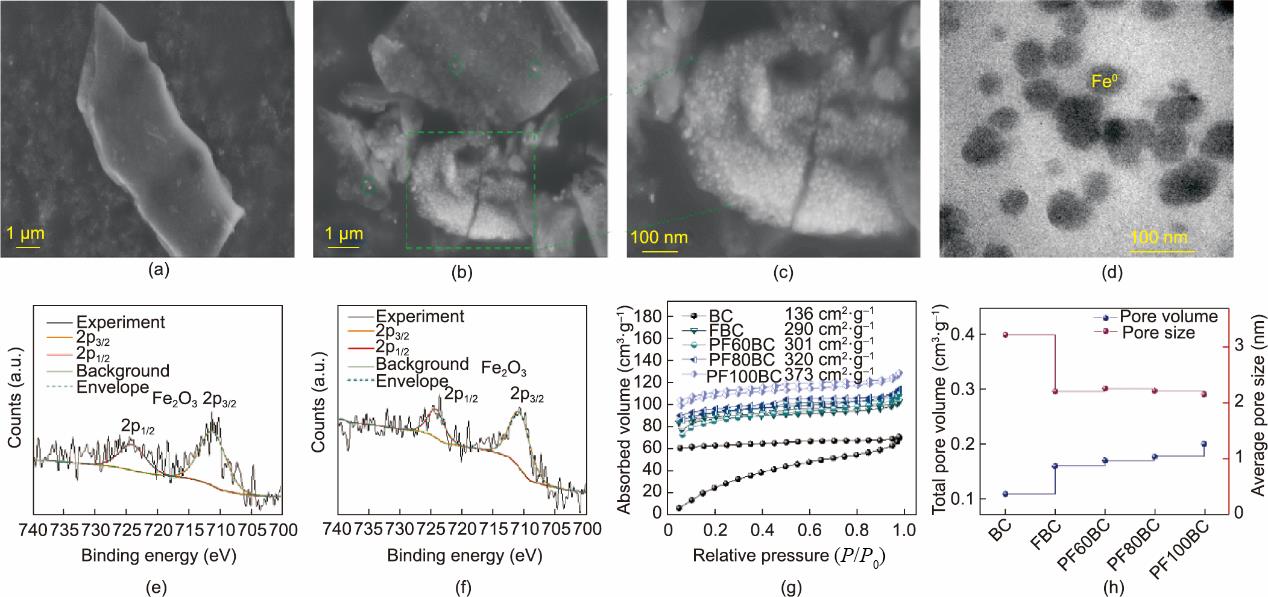
图2 FBC(a)和PF100BC(b, c)的SEM图;(d)PFBC的TEM图;FBC(e)和PF100BC(f)的高分辨Fe2p光谱;(g)所得材料的N2吸/脱附曲线;(h)所得材料的孔容和孔径。
(5)XPS分析。所得生物炭的表面元素组成采用XPS检测,结果列于附录A中的表S1,图2(e)、(f)以及附录A中的图S2(b)中。Fe的含量从BC(0.39%)增加到PF100BC(0.93%);该结果与XRD和VSM结果一致。此外,FBC和PF100BC的光谱中在约711 eV和约724 eV处存在两个典型卫星峰[图2(e)~(f)],该峰属于Fe2O3的2p3/2和2p1/2卫星峰[33‒34]。因此,附着在FBC和PF100BC表面的物质被判断为Fe2O3。该化合物可能是由Fe2O3的不完全还原产生的。此外,BC中Fe的原子百分比等于FBC中Fe的原子百分比,但Fe2O3的信号峰没有出现在BC中[图S2(b)],其可能是来源于生物质RS表面原有的铁。基于以上分析,所制备的nZVI-BC由生物炭内部的Fe0和表面少量的Fe2O3组成。
(6)BET分析。N2吸脱附等温线及孔容和孔径的信息如图2(g)、(h)以及附录A中的表S2所示。通过分析XRD、SEM和TEM得出结论,随着Fe0含量的增加,比表面积(SBET)增加,孔径(Dp)减小,这可能归因于nZVI颗粒占据了生物炭的内部[26]。
与BC和FBC相比,PFxBC的SBET和Vtot分别从136 m2·g-1和290 m2·g-1增加到373 m2·g-1,从0.1100 cm3·g-1和0.1610 cm3·g-1增加到0.2015 cm3·g-1,表明Fe0分散在生物炭内部。此外,所得生物炭的Dp具有较小的介孔(2~4 nm),而BC中不含微孔。然而,由于RS的改性,随着Fe0的引入,微孔体积(Vmicro)增加,表明一些小孔被Fe0颗粒堵塞。
综上所述,本文的改性方法可以成功地制备出nZVI-BC,并且合成材料具有优异的特性。与nZVI-BC的其他制备方法(表1 [7‒9,35‒36])相比,该方法具有步骤简单、时间短、成本低、环境友好且无需添加额外还原剂的优点。
《表1》
表1 nZVI-BC制备方法的比较
| Name | Precursor | Preparation method | Detailed Procedures | Reference |
|---|---|---|---|---|
| nZVI@BC | Cornstalk | Liquid reduction | Pyrolysis at 500 °C for 2 h; soak in FeCl3·6H2O for 24 h; purge N2 for 30 min; NaBH4 reduction for 30 min | [ |
| C-Fe0 | Carbon black | Carbonthermal reduction | Fe(NO3)3·9H2O adsorption; vacuum dry; pyrolysis at 800 °C for 3 h under Ar atmosphere | [ |
| biochar-supportednZVI | Rice straw | Liquid reduction |
Pyrolysis at 500 °C, 700 °C; mix with FeSO4·7H2O to agitate for 24 h; purge N2 for 1 h; KBH4 reduction |
[ |
| Fe/C composites | Maize cob | Liquid reduction |
Pyrolysis at 600 °C for 2 h; mix with FeSO4·7H2O to shake for 12 h; NaBH4 reduction for 1 h under N2 atmosphere |
[ |
| Fe0@C | Glucose monohydrate | Carbonthermal reduction | Hydrothermal treatment using Fe3O4 at 180 °C for 10 h; pyrolysis at 700 °C for 2 h | [ |
| nZVI-BC | Rice straw | Carbonthermal reduction | Heat treatment using FeCl3·6H2O and PEG400 at 80°C for 30 min; pyrolysis at 700 °C for 2 h | This study |
除此之外,如附录A中的图S3所示,PF100BC的稳定性通过在不同pH值下的zeta电位和不同时间的XRD谱图被证明。zeta电位结果表明PF100BC在很宽的pH范围内具有优异的稳定性。当pH > 6时,材料表面带有大量负电荷。XRD结果表明材料在240 d内表现出高稳定性。Fe0的典型衍射峰强度与初始材料的Fe0的衍射峰强度相匹配。结果中未观察到其他氧化物质。
除了nZVI-BC的物理性质外,其化学结构也同样重要。图3显示了通过FTIR和XPS获得的材料上化学官能团和化学元素种类的信息。
《图3》

图3 (a)BC、PF80BC和PF100BC的红外光谱;(b)BC的高分辨O1s光谱;(c)PF80BC的高分辨O1s光谱;(d)PF100BC的高分辨O1s光谱。
《3.1.2. 化学特性》
3.1.2. 化学特性
(1)FTIR分析。如图3(a)所示,BC中的主要谱带如下:3433 cm-1处的强吸收峰代表了材料表面的O‒H的伸缩振动,1617 cm-1位置是芳香环中的C=C和C=O的伸缩振动峰[37],1400 cm-1处的强吸收峰表示存在‒COO或内酯结构[38],1091 cm-1处的强吸收峰代表C‒O‒C的伸缩振动,1049 cm-1、801 cm-1和880 cm-1处的吸收峰分别对应于Si‒O‒Si的非对称振动和对称振动,460 cm-1处的吸收峰是由于O‒Si‒O的弯曲形成的[39]。
以往的报道中表示BC中存在有机和无机官能团[40]。然而,nZVI-BC的FTIR光谱显示结果与之不同。PF80BC和PF100BC在O‒H和Si‒O‒Si的位置显示出明显的变化以及在C=C位置显示出微小的变化。基于PF80BC和PF100BC中ZVI的嵌入,其占据了生物炭上的一些活性位点。有文献表明,Si‒O‒Si峰在880 cm-1和1049 cm-1处的显著降低或消除,表明Fe0和Si‒O‒Si之间发生了反应[36]。O‒H从3428 cm-1偏移至3445 cm-1,这种转变可归因于铁的附着[39]。此外,在1617 cm-1处出现的代表芳香环上的C=C和C=O峰由宽而不规则的强峰变为窄的强峰。这可能是来源于RS的木质素组分经高温热解形成的,因为纤维素和侧链中的脂肪烷烃发生了解构[37]。
FTIR分析初步表明,嵌入生物炭内部的Fe0以Si‒O‒Fe的形式键合。nZVI-BC具有强的C=C骨架,将Fe0颗粒分散在芳环结构内,避免了Fe0的聚集和氧化。为了进一步确定nZVI-BC中的Fe0的连接形式,BC和PFxBC表面氧形态的变化通过XPS检测。
(2)XPS分析。对XPS O1s峰进行分峰拟合得到四个峰,分别是位于约532.9 eV、约533 eV、约531 eV和约530 eV处的SiO2、C‒O、金属碳酸盐和金属氧化物[41]。显然,BC中的氧元素主要来源于SiO2 [图3(b)],其相对含量为 51.7%。该结果也可以通过BC的XRD分析来解释(附录A中的图S4)。与其他样品相比,SiO2的特征峰仅出现在BC的XRD图中,表明BC中SiO2含量较高。PF80BC和PF100BC中的SiO2占比较低,分别为43.75%和24.35% [图3(c)和(d)]。正如FTIR的分析,PF80BC和PF100BC中的SiO2含量降低是由于Fe0颗粒的产生并与SiO2反应造成的。此外,P80FBC和PF100BC中C‒O含量相较于BC中的37.84%提高到47.90%和67.06%,其成为这两种生物炭中氧的主要存在形式。XPS的分析结果确认了本文制备的零价铁生物炭化学组成Si‒O‒Si和C‒O含量的变化,并证明了生物炭表面存在氧化铁[图3(c)和(d)]。
根据以上的分析结果得出结论,nZVI-BC由表面的Fe‒O (Fe2O3)和S‒O (SiO2)以及壳核内的ZVI和C=C骨架组成。这种“核-壳”结构有利于保护Fe0免受氧化。
《3.2 RS改性机制》
3.2 RS改性机制
根据所得生物炭的特性分析表明,PEG400的加入增强了单一的FeCl3对RS的改性。为了探索具体的强化机制,改性前后的RS的理化结构变化被表征。
《3.2.1. RS结构变化》
3.2.1. RS结构变化
从图4(a)~(c)可以看出,未改性RS的表面由于木质素的存在表现为坚硬而平滑的特性。RS经酸性FeCl3改性后,木质素以液滴的形式重新排列沉积在F-RS的表面[36]。然而,PEG400被引入后,PF80-RS的结构变得松散,木质素在溶剂分解作用下被分离[42‒43]。
《图4》

图4 RS(a)、F-RS(b)和PF80-RS(c)的SEM;RS(d)、F-RS(e)和PF80-RS(f)的EDS元素分布图;RS(g)和F-RS(h)和PF80-RS(i)的EDS能谱图。
此外,不同改性引起的RS变化也反映在其表面元素的颜色和分布上。从附录A中的图S5可以看出,未改性的RS为黄色,F-RS变为黑褐色,PF80-RS和PF100-RS则为红褐色;这种颜色差异可能是由于残留在RS表面的木质素或木质素和氧化铁的混合物造成的。在先前的报道[44‒45]中提到过生物质在高温高压下通过FeCl3预处理,表现出脱木质素的作用,相应的木质素官能团信号减弱。不过在本研究中,FeCl3处理条件为常压,处理温度不超过100 °C。因此,如FTIR结果(附录A中的图S6)所示,单一的FeCl3处理不会导致木质素的大量去除,其化学结构未发生明显变化。
另外,EDS能谱和元素分布扫描图[图4(d)~(i)]显示F-RS和PF80-RS表面的Fe含量从未处理的RS的0.1%分别增加到0.3%和2.0%,表明了PEG400的添加有利于改性RS表面的铁相沉积。通过XPS确认改性RS表面上沉积的铁来源于Fe2O3(图5)。因为在所得Fe2P峰中观察到两个典型的卫星峰出现在约711 eV和约724 eV的位置,它们属于Fe2O3的2p3/2和2p1/2卫星峰。并且,PF80-RS的峰强明显高于F-RS。PF80-RS表面Fe的原子含量为3.33%,比F-RS(0.32%)高9.4倍。该结果与EDS能谱的结果一致。此外,更多的Fe(5.47%)出现在PF100-RS表面(附录A中的表S3),说明了改性温度的升高有利于FeCl3的转化。
《图5》
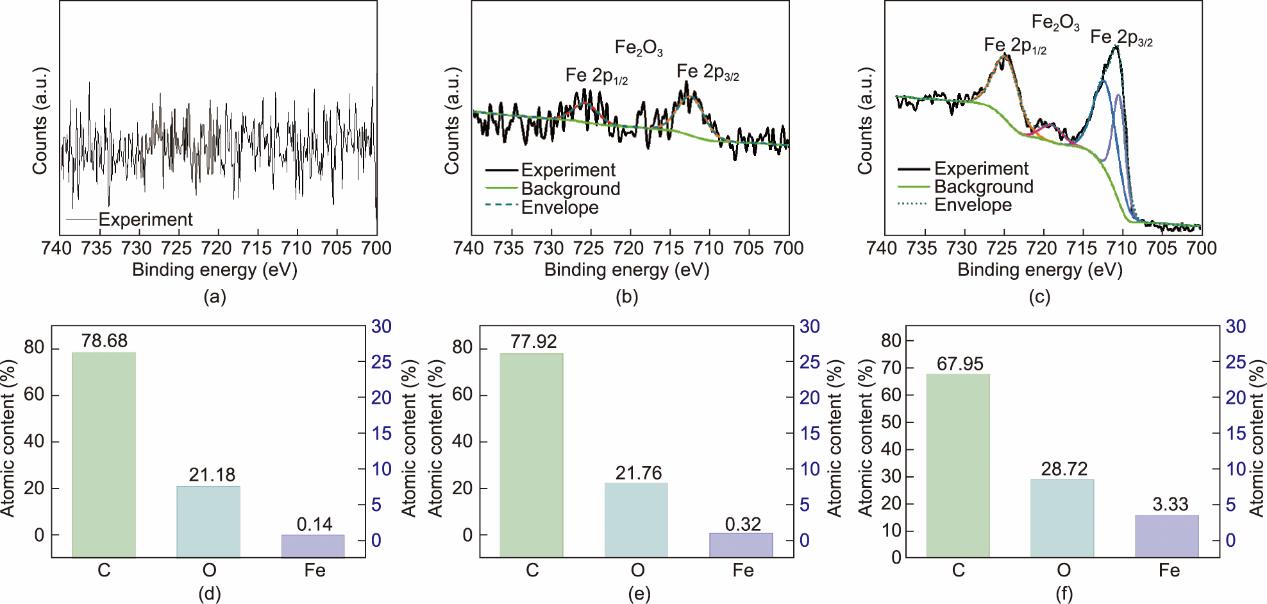
图5 RS(a)、F-RS(b)和PF80-RS(c)的高分辨Fe2p光谱;RS(d)、F-RS(e)和PF80-RS(f)的元素原子含量。
因此,与单独FeCl3改性相比,PEG400的添加起到了改善RS的结构变化及促进Fe2O3产生的双重作用。
《3.2.2. 铁相转移机制》
3.2.2. 铁相转移机制
PEG是一种两端具有活性‒OH基团的两亲性聚合物[46],它通常被认为是一种相转移催化剂[47]。在液相反应系统中,随着PEG的增加,质子的数量随之减少[48]。在FeCl3改性的RS体系中,FeCl3因加热发生水解。产生的Fe(OH)3沉积在RS上,在样品干燥后转化为Fe2O3,此过程发生的反应如反应方程式(2)和(3)所示。
(2)
(3)
然而,FeCl3在此条件下的水解作用可能较弱,且受到生成的H+的抑制。因此,在F-RS中检测到少量Fe2O3 [图4(h)和图5(e)]。添加PEG400和升高处理温度后增大了H+的消耗[48],促进了水解反应,产生更多的Fe2O3 [图4(i)和图5(f)]。
除此之外,PEG400的引入带来的RS表面结构的变化对Fe2O3的附着发挥重要作用。FeCl3改性后的F-RS表面仍然是平滑坚硬的[图4(b)],不利于Fe(OH)3的吸附。但是PF-RS的表面却是粗糙的,这可能为吸附Fe(OH)3提供了便利,使干燥后的PF-RS表面被检测到更多的Fe2O3。
综上所述,PEG400的增加改善了改性RS的表面结构,促进了FeCl3的水解,使更多的Fe2O3积累在生物质表面。我们称这种改性为“铁相转移”,其可能的机制如图6所示。该结果对于后续的热解生产nZVI十分重要。
《图6》

图6 PEG400促进FeCl3改性RS的铁相转移机制。
《3.3 nZVI-BC合成的热解机制》
3.3 nZVI-BC合成的热解机制
热解过程中,Fe0的形成主要归因于生物质热解产生的碳和还原性气体(CO和H2)[49]。生物质热解释放的挥发物在还原过程中起着重要作用[50],在这些还原气体的帮助下,氧化铁被还原为Fe0。还原气体的产生取决于热解温度,其种类和产量影响着还原反应的质量[51]。根据之前的研究,我们知道在整个热解范围内,CO的产生主要来自于半纤维素的分解,由纤维素热解产生的气体量相对较少,而木质素产生的气体主要发生在高于600 ℃的温度下[17]。CH4主要来源于500~580 ℃的木质素的C‒C断裂,H2主要是随着温度的升高从C‒H键断裂中产生的[52]。
PF100-RS和PF100BC的FTIR结果如图7所示,吸收峰的分配见附录A中的表S4 [15,53‒54]。1384 cm-1、1245 cm-1、1163 cm-1、1061 cm-1和898 cm-1处代表纤维素和半纤维素的主要吸收峰消失了,表明C‒H、C‒O和C‒O‒C键的断裂。与木质素的芳香骨架振动相关的吸收峰仅在1513 cm-1处消失,表明C‒C键的断裂并不容易。在1732 cm-1处的C‒O和C=O键断裂主要引起CO和CO2的生成。H2则是由在2919 cm-1和1384 cm-1处的C‒H断裂产生的。此外,有文献表明一些C‒C键的断裂会导致少量的CH4生成,并且气体产量随温度的升高而增加[18]。在这些气体的作用下,Fe2O3被还原为Fe0。PEG400不仅增加了Fe2O3的产生并改善了RS表面结构,同时保证了较多的木质素被保留于改性RS中。所以,热解改性RS几乎可以提供所需的所有还原气体。此外,通过CO和H2还原Fe2O3生成Fe0的过程中会产生两种中间体——Fe3O4和FeO(温度高于570 °C)[14]。在特定的热解温度下可以产生充分的还原气体作用于Fe2O3,使其被还原为Fe0。Fe0的形成可由反应方程式(4)~(9)解释。
(4)
(5)
(6)
(7)
(8)
(9)
《图7》

图7 PF100-RS和PF100BC的FTIR光谱图。
此外,为了研究Fe2O3是否可以在较低的热解温度下还原为Fe0,我们对使用PF80-RS和PF100-RS在500 ℃下制备的材料进行比较。XRD结果(附录A中的图S7)显示在约44.7°处没有出现Fe0的衍射峰,表明500 ℃热解释放的气体不足以将Fe2O3完全还原为Fe0。一些报道表明,随着温度升高,CO、CH4和H2的产量会增加[51]。因此,若想通过碳热还原获得零价铁形式的生物炭,合适的热解温度至关重要,这一结果与之前的研究一致[49]。
热解温度低(500 ℃)除了造成气体产量低以外,在热解过程中产生的固体碳也发挥着一定的作用。已有报道证明还原铁的反应发生在气相(如CO和H2)中,而不是在固体碳材料上[55]。但是,碳可以与CO2发生反应[反应方程式(10)]造成CO2的减少。
(10)
在热解过程中,CO2主要产生于低温(< 500 ℃)下半纤维素的热解和高温(> 500 °C)下木质素的热解[17]。如XPS光谱结果(附录A中的图S8和表S5)所示,PF80BC和PF100BC的O/C比分别从0.120和0.130(500 ℃)降低到0.110和0.086(700 ℃)。而PF80BC和PF100BC的sp2-C(约284.4 eV)的相对含量占比分别从36.19%(500 ℃)升高到47.77%(700 ℃),从36.31%(500 ℃)升高到55.68%(700 ℃);sp3-C(约285.0 eV)的相对含量占比则分别从38.19%(500 ℃)减少到32.94%(700 ℃),从37.25%(500 ℃)减少到27.72%(700 ℃),说明了nZVI-BC的石墨化程度得到改善[56]。这个结果可能是由无定形碳参与CO2的还原导致的;根据反应方程式(4)~(6),所产生的CO将继续参与Fe2O3的还原反应。
本研究通过原位碳热还原获得Fe0,减少温室气体排放和环境污染;这种nZVI-BC的制备方法可被看作一种绿色生产工艺。nZVI-BC的形成机制如图8所示。
《图8》
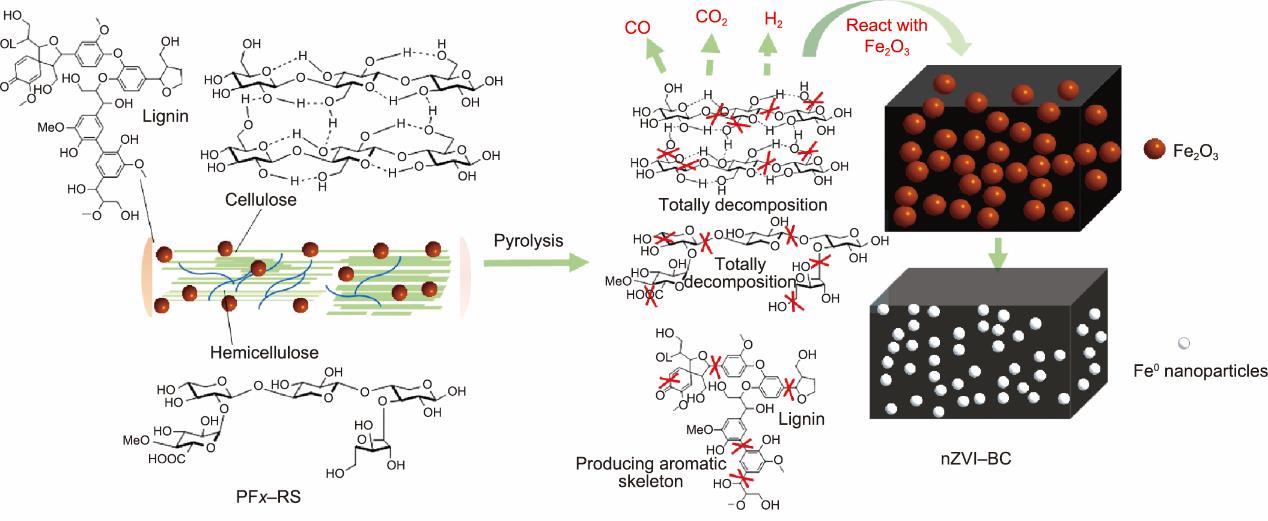
图8 合成nZVI-BC的热解过程图解。
《3.4 nZVI-BC在CR去除中的应用试验》
3.4 nZVI-BC在CR去除中的应用试验
为了确认PF100BC(nZVI-BC)去除污染物的能力,我们使用PF100BC作为催化剂进行了高级催化氧化去除CR的实验。另外还进行了单独的PS氧化和PF100BC吸附的对照实验。
《3.4.1. nZVI-BC应用于刚果红的去除实验》
3.4.1. nZVI-BC应用于刚果红的去除实验
从图9(a)和(b)可以看出,与单独使用PS氧化相比,PF100BC对CR的去除率大幅度增加。在相同的监测时间内,使用PF100BC作为催化剂处理的CR剩余浓度远低于未使用PF100BC的情况,表明PF100BC具有良好的催化高级氧化能力。在5 min内,PF100BC表现出70.6%的高效吸附能力;在相同时间内的催化降解效率为75.67%。PS氧化对CR的去除率仅为4.3%。在PF100BC的催化下,CR的氧化效率提高了17.6倍。在60 min时,CR的催化氧化去除率达到90%,剩余浓度为3.0 mg·L-1。此时的PF100BC对CR的吸附去除率仅为61.3%,这可能是由于不稳定的吸附导致脱附造成的。PS对CR的氧化效率仅为8.3%,表明在没有nZVI催化的情况下,PS的氧化反应缓慢。根据附录A中的图S9所示,PF100BC对CR的催化降解符合伪一级动力学[57]。
《图9》

图9 CR的催化氧化去除实验。(a)~(c)PS氧化、nZVI-BC吸附和nZVI-BC催化氧化降解CR的过程图、去除效率和紫外扫描图;(d)、(e)nZVI-BC使用前后的XRD和FTIR光谱;(f)催化降解CR后的nZVI-BC的EDS元素分布扫描图。实验条件:CR的浓度为30 mg·L-1,投加量为0.5 g·L-1,PS为1.8 mmol·L-1,温度为30 ℃。
此外,对三个不同体系下CR去除后的上清液进行波长扫描[图9(c)],结果显示,经PS氧化后的CR在498 nm处仍有明显的吸收峰;经过nZVI-BC吸附后的CR溶液的显色吸收峰强度减弱但没有消失;加入PF100BC催化降解后的上清液中CR的显色峰完全消失,表明CR上偶氮发色团的断裂可能与nZVI有关[58]。
《3.4.2. nZVI-BC对CR的去除机制》
3.4.2. nZVI-BC对CR的去除机制
为了探索PF100BC对CR的去除机制,我们通过XRD、FTIR及EDS对使用前后的PF100BC进行了检测。XRD谱图[图9(d)]显示使用前后的PF100BC几乎没有变化,使用后只有ZVI的峰值下降,这可归因于ZVI和S2O
(11)
(12)
FTIR光谱[图9(e)]结果显示‒COO和‒OH的吸收峰在吸附和催化氧化反应后消失。这种消失可能是由于‒COO和‒OH基团与染料上的‒NH2形成了氢键造成的[59]。从不稳定的吸附结果和增强的高级催化氧化的结果来看,CR的去除可以解释为:一方面是依靠官能团的键合作用被PF100BC吸附;另一方面被Fe0活化S2O
总之,通过本研究所述方法制备的PF100BC显示出了高效的高级氧化催化能力。后期我们将继续研究该材料对CR去除的最佳应用条件,如投加量、CR初始浓度和pH影响。同时详细地研究降解机制,包括电子转移机制、自由基作用和污染物降解产物等内容。
《4、 结论》
4、 结论
本研究通过将PEG400引入FeCl3改性RS的反应体系,利用碳热还原制备出了磁性nZVI-BC。在RS改性中,PEG400的添加促进了FeCl3的水解,改善了RS表面结构,增加了Fe2O3在RS表面的附着。在热解过程中,碳化的RS和Fe2O3在还原气体的参与下发生氧化还原反应,形成nZVI-BC。该方法制备的nZVI-BC物相纯净,Fe0嵌入在碳骨架内,不易被氧化。在nZVI-BC用于CR的催化高级氧化测试中,5 min时其表现出70.6%的快速吸附;在60 min时实现了90%的污染物去除率。本研究中提出的新型改性方法过程简单、成本低廉、用时较短。
该研究对于指导生物质向生物炭的转化具有重大意义,并且具有推动nZVI-BC规模化生产的巨大潜力,有望在未来实现批量生产并应用于环境修复。











 京公网安备 11010502051620号
京公网安备 11010502051620号




