
《1. 引言》
1. 引言
单克隆抗体(mAb)一直是治疗许多疾病的重要选择,并有望在未来的相关疾病治疗中发挥重要作用[1-2]。抗体药物的研发始于小鼠杂交瘤细胞;然而,早期将鼠源抗体作为治疗药物的尝试一波三折,主要是因为鼠源抗体会引起明显的免疫反应,效应功能较弱。用于制备嵌合或人源化单克隆抗体的新技术有效缓解了这些问题。与亲代鼠源抗体相比,作用相似或具有更好抗原结合功能的嵌合/人源化单克隆抗体具有较弱的免疫原性与更低的细胞毒性,预示着这些抗体具有更好的治疗效果。20世纪90年代以来,抗体药物在慢性炎症、肿瘤、传染病、移植和心血管疾病等临床应用中被视作一类新的治疗选择。然而,开发嵌合或人源化抗体却具有挑战性。嵌合和人源化均可导致抗体亲和力和(或)生物活性的降低,而确保结合亲和力和生物活性的一致性则较为耗时且成本较高。此外,嵌合和人源化单克隆抗体的其他非人源部位仍可引起免疫原性反应,尤其是3~5年后。
用于制备全人单克隆抗体的新技术的出现,减少了对嵌合/人源化抗体的需求。理论上,全人抗体应该是最安全的,并且具有最小的免疫原性。目前进入临床试验的抗体大多是人源化或全人抗的,这也被认为是抗体制剂的发展趋势。
目前有如下几种技术可用于制备全人抗体:噬菌体展示技术(可用于构建全人抗体库)、人-人杂交瘤技术、转基因小鼠技术、B淋巴细胞永生化和克隆技术以及单细胞聚合酶链反应(PCR)。目前大多数正在开发的全人抗体是在体外通过噬菌体展示技术或在体内通过含有人抗体基因的转基因小鼠产生的。通过噬菌体展示技术可筛选出全人抗体,不需要进行动物免疫。几种通过噬菌体展示技术获得的全人抗体药物正处于临床试验的不同阶段。2003年1月,修美乐(阿达木单抗)作为雅培推出的第一个从噬菌体库中筛选出的全人抗体药物被批准上市。目前,该药物已被批准用于治疗多种疾病,包括类风湿性关节炎、斑块状银屑病和克罗恩病等。然而,在修美乐研发中心发现,从抗体库中进行第一轮筛选获得的噬菌体抗体都表现出了较弱的结合亲和力,因此对于亲和力的增强有必要开展更为深入的研究;通过转基因小鼠获得的全人抗体的亲和力通常是足够高的,提示在加强免疫过程中发生了抗体体内亲和力成熟。正常情况下,高亲和力抗体并不需要通过增强体外亲和力或使用其他技术增强单克隆抗体效价。2006年,抑制表皮生长因子受体信号通路的帕尼单克隆抗体成为第一个来源于转基因小鼠的单克隆抗体药物。目前,也已有一系列由转基因小鼠产生的全人抗体被报道,它们是进入临床试验的良好候选者。
在最近的许多临床试验中,低毒性和高疗效的抗体候选物越来越成为加速新一代药物开发关注的焦点,但获得治疗性抗体相当耗时。通过结构的设计来增强抗体药物活性的方法是有效的,如提高亲和力[3-4]、效应生物功能[5]以及物理和生化稳定性[6-7]等。
计算机辅助同源建模的方法为抗体药物的开发做出巨大贡献,尤其是在人源化抗体开发的早期阶段。这种建模方法的整体潜力目前尚不明确,且与小分子药物发现中使用的计算方法相比,这种建模方法还没有得到很好的发展。基于当前对抗体机制的理解深度和结构分辨技术水平,我们还是有理由相信通过计算机辅助分子设计方法设计新型抗体分子是可能的。在前期工作中,我们已经构建了包括肽-Fc融合蛋白和单结构域抗体在内的一系列功能性抗体模拟分子[8-11]。
本文借助基于结构的计算机辅助抗体设计技术,描述了一种靶向人表皮生长因子受体2(HER2/erb-B2)的新型全人抗体。HER2/erb-B2在20%~30%的浸润性乳腺癌中过度表达,这也使其被确定为癌症的重要标志物[12]。几种靶向HER2的抗体,如赫赛汀(曲妥珠单抗),在乳腺癌患者的体内和体外试验中均显示出了有效的抗肿瘤活性[13-14]。X射线晶体学和分子模型提供了HER2与赫赛汀之间的相互作用模式[15]。基于这些信息,我们确定了赫赛汀识别HER2的潜在表位,并利用抗体设计方法获得了一种名为“HF”的新型抗HER2全人单克隆抗体,该抗体具有良好的抗肿瘤活性。
《2. 材料和方法》
2. 材料和方法
《2.1. 试剂》
2.1. 试剂
HER2蛋白(目录号:10818-H08H)购自中国Sino Biological公司;人IgG同型对照(目录号:02-7102)购自美国Invitrogen公司;藻红蛋白(PE)结合的山羊抗人多克隆抗体(PE_GAH)(目录号:12-4998-82)购自美国eBiosciences公司;DMEM培养基(目录号:11965-092)和胎牛血清(FBS;目录号:10438-034)购自美国Thermo Fisher公司;DELFIA EuTDA细胞毒性试剂(目录号:AD0116)购自美国Perkin Elmer;其他所有试剂均为分析级。
《2.2. 细胞系》
2.2. 细胞系
人乳腺癌细胞系SKBR3(ATCC® HTB-30)、BT474(ATCC® HTB-20)、MCF-7(ATCC® HTB-22)、T47D(ATCC® HTB-133),人卵巢癌细胞系SKOV3(ATCC® HTB-77)、人肝细胞癌(HepG2, ATCC® HTB-8065)和人胚肾上皮细胞293T(ATCC® CRL-3216)购自美国典型培养物保藏中心(ATCC)。使用Ficoll-Paque PLUS(目录号:17-1440-02;GE Healthcare, USA)从全血中分离外周血单核细胞(PBMC)。在DMEM培养基(目录号:11995-500;Gibco, USA)中添加10%热灭活胎牛血清(目录号:26010-074;Gibco, USA)、100 U·mL-1青霉素和100 U·mL-1链霉素(目录号:15140-122;Gibco, USA),用于在37 ℃下、5% CO2中培养细胞。使用LookOut支原体PCR检测试剂盒(目录号:MP0035;Sigma-Aldrich,USA)检测细胞,以确认其无支原体污染。
《2.3. 计算机辅助人源化和表位预测》
2.3. 计算机辅助人源化和表位预测
使用InsightII软件(2005版,Molecular Simulations公司,USA),通过计算机辅助同源建模的方法构建HER2和抗体的三维(3D)结构。通过分子对接构建HER2和抗体的3D复合物结构并分析其潜在表位,使用InsightII软件中的Discover和CHARMM模块进行优化。非键合截止值为10 Å,非键合参数和原子电荷取默认值。在真空计算中使用距离相关介电常数。分别使用最陡下降(2000步)和共轭梯度(5000步)方法将模型最小化。
基于表位,设计6个多肽,并将其设置为合适的框架区(FR)中的6个互补决定区(CDR),由此形成一种新抗体HF。然后从潜在表位、亲和力等方面分析HER2/HF复合物的理论结构。
《2.4. 抗体依赖性细胞毒性》
2.4. 抗体依赖性细胞毒性
根据生产商说明,使用DELFIA EuTDA细胞毒性试剂(PerkinElmer, USA)检测抗体的细胞毒性试验活性。简言之,将靶细胞(如肿瘤细胞)与荧光增强配体共同孵育,再与稀释的抗体在37 ℃下孵育1 h,然后加入效应细胞(如PBMC)。效靶比为50∶1。37 ℃下再孵育4 h后,采用时间分辨荧光分析仪测定上清液荧光。使用在0.2% Triton X-100(Gibco, USA)中裂解的细胞样本测定最大释放量。通过以下公式计算各单克隆抗体处理样本中的裂解百分数。
《2.5. 生物分子相互作用分析》
2.5. 生物分子相互作用分析
将市售抗原HER2蛋白设定为液相,将赫赛汀设定为阳性对照。将溶剂相(即样品缓冲液)设定为阴性对照。根据V基线扣除背景,以便获得理想的动力学曲线,并计算解离常数(KD)值。用于计算摩尔浓度的HF和赫赛汀分子量均取148 kDa。
《2.6. 流式细胞术》
2.6. 流式细胞术
在冰上进行实验。先清洗细胞,然后用一系列稀释的单克隆抗体或免疫球蛋白G(IgG)同型对照孵育。用PE_GAH清洗和孵育后,使用BD FACSCalibur(BD Biosciences, USA)测定细胞样本,并通过BD CellQuest Pro软件(BD Biosciences, USA)分析荧光密度。
在表位鉴定试验中,采用丙氨酸置换法合成一系列HER2突变体,并在293T细胞表面表达。接下来,用抗体HF作为一抗孵育细胞,然后用PE_GAH作为二抗进行检测。
《2.7. 小鼠体内试验》
2.7. 小鼠体内试验
8~10周龄雌性BALB/c裸鼠共18只,购自北京维通利华实验动物技术有限公司。将小鼠饲养于无特异病原体(SPF)级动物房中,每笼3只,喂食无菌水和食物。在体内试验期间,我们在小鼠的使用方面遵循了《动物研究:体内实验报告指南》(Animal Research: Reporting of In Vioo Experiments,ARRIVE)指导原则,并根据3R原则(即替代、减少和优化)观察动物伦理学。小鼠的使用或治疗完全符合研究动物使用指南,并经北京毒物药物研究所动物伦理委员会批准。检查小鼠的不适性和总体外观。研究结束后,小鼠在麻醉状态下死亡,然后对其进行颈椎脱臼手术;我们使用深度冷冻技术确认其死亡。
对于异种移植模型,对每只小鼠皮下注射2 × 106个肿瘤细胞。一周后,将小鼠随机分为3组,每组6只,静脉给予人IgG同型对照或单克隆抗体,并在给予抗体后每周观察两次,持续4周。第28天观察并处死小鼠,然后分离器官进行成像。抗体给药前先腹腔注射80 mg·kg-1氯胺酮、20 mg·kg-1甲苯噻嗪、0.6 mg·kg-1阿托品混合液以麻醉小鼠。使用VersArray 1300B设备检测器(Roper Scientific, the Netherlands)对每只小鼠进行成像,并在暴露5 min后对光信号进行测定。实验进行两次。采用MetaVue软件(Molecular Devices,USA)进行数据分析。
《2.8. 统计学分析》
2.8. 统计学分析
所有实验都至少进行两次。数据均呈现为平均值±标准差的形式。比较几组之间的成对差异。通过t检验或单因素方差分析(ANOVA)的重复测量进行统计分析。当P值小于0.05时认为具有统计学显著性。
《2.9. 伦理批准》
2.9. 伦理批准
小鼠的使用或治疗完全符合研究动物使用指南,并经北京毒物药物研究所动物伦理委员会批准。
《3. 结果》
3. 结果
《3.1. 通过计算机辅助设计方法设计的新型全人抗体HF》
3.1. 通过计算机辅助设计方法设计的新型全人抗体HF
新型全人抗体设计方案包括5个主要步骤:①根据抗原和抗原-抗体复合物(即HER2-赫赛汀)的3D结构确定抗原(即HER2)的功能表位,并利用生物定点诱变进行分析。②基于功能表位的表面理化性质,考虑潜在关键残基与表位的相互作用模式(如分子间氢键、静电和范德华相互作用),筛选和定向关键氨基酸。计算出合适的距离和角度。为确保潜在关键残基的合适位置,引入甘氨酸、丝氨酸和丙氨酸作为连接体。③根据免疫球蛋白(IGV)文库可变区中免疫球蛋白重链可变区(IGHV)/免疫球蛋白κ链可变区(IGKV)的CDR的同源性,选择全人抗体重链可变区IGHV3-66和轻链可变区IGKV1-39作为支架,对筛选出的关键氨基酸进行优化展示,进而实现虚拟亲和成熟。基于潜在的3D支架结构(即IGHV3-66和IGKV1-39的框架区域),在6个CDR中替换了步骤②中设计的合适肽。④基于设计的抗体序列,构建可变片段(Fv)的3D结构,包括重链(HFv)和轻链(LFv)的Fv。逐一分析Fv片段的取向和构象,保持Fv片段的稳定构象,进行深入研究。⑤利用计算机辅助同源建模方法构建新型抗体可变结构域LFv和HFv的3D结构。评估LFv与HFv的结构互补性,采用分子对接方法构建Fv片段的3D结构。此外,使用分子对接方法构建HER2-Fv复杂模型,并使用分子动力学进行优化。以结合能和稳定能为标准,选择与HER2结合能最低的抗体Fv片段进行检测。
为了确定HER2蛋白功能结构域的分子构象信息,我们分析了HER2-赫赛汀的3D晶体复合物(蛋白数据库代码:1n8z),并预测了HER2的关键表位[图1(a)]。利用溶剂可及表面积计和表面静电分布等方法考察HER2和赫赛汀之间的相互作用[15],确定了人HER2胞外域C端的关键残基[图1(b)]。残基Y560、K561、D562和F565(在本文中命名为D1)是赫赛汀识别的表位。由于残基K585和I583(命名为D2)比D1具有更好的可及性和更好的正电势,因此被预测为首要的潜在关键表位。研究中使用命名为D3的结构域(包括D552、Q553和E550)作为比较。实验结果表明理论结果是合理的[图1(c)]。由于残基582~585靠近膜并接近重要表位(即赫赛汀鉴定的HER2的560~565),因此这些近膜残基可能是HER2的另一个关键结构域。
《图1》

图1 人HER2胞外域的3D结构和表位预测。(a)根据HER2的晶体结构,基于计算机辅助同源建模方法的HER2的3D理论带状结构;(b)HER2 C端潜在的关键氨基酸残基;(c)HER2 C端潜在关键氨基酸残基的方向。
HER2中残基K561和K585形成正电位位置,残基D562形成负电位位置;残基F565和I583形成疏水核心,而残基D562、K585、K561和Y560形成亲水核心。此外,残基F565和Y560中的侧链芳环形成了
使用计算机辅助分子设计方法选择和定向关键残基,该方法考虑了氨基酸潜在的分子间相互作用(如疏水相互作用,
《图2》
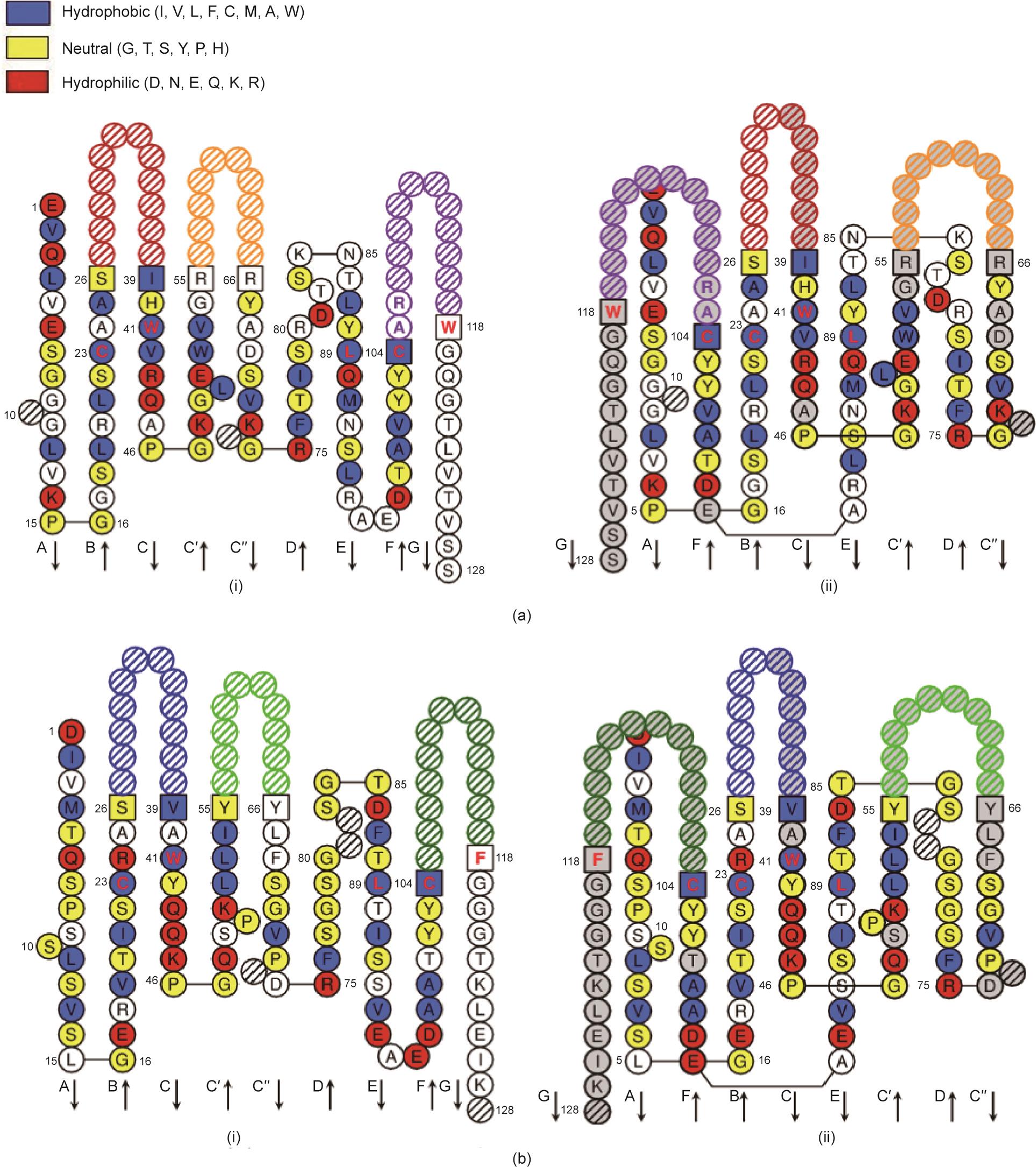
图2 新型抗HER2抗体HF的设计。(a)使用Collier-de-Perles工具分析所选重链可变区域框架的拓扑结构:(i)单层拓扑结构;(ii)双层拓扑结构。(b)使用Collier-de-Perles工具分析所选轻链可变区域框架的拓扑结构:(i)单层拓扑结构;(ii)双层拓扑结构。
利用计算机辅助同源建模和分子力学优化方法,构建新型全人抗体的3D结构。根据Fv的稳定能量以及重链可变区(VH)与轻链可变区(VL)之间的相互作用,分析CDR中所选关键残基的构象和方向,并选择合适的抗体。
根据HER2及其功能性抗体赫赛汀的结构,使用计算机辅助分子对接方法对HER2的结构[图1(a)]和所设计的候选全人抗体进行结构模拟,并分析相互作用能、分子间氢键和疏水相互作用。最后,选择名为HF的抗体,它与上述关键表位的结合能力更强、作用能更低、复合物更稳定,因此性能可能更好。
HF的3D结构如图3(a)所示。由4个CDR(即HCDR1、HCDR2、LCDR1和LCDR2)形成的亲水核心包裹着CDR3区(即HCDR3和LCDR3)。LCDR3中的His91和Tyr92、HCDR1中的Phe27,以及HCDR3中的Phe104残基形成疏水核并具有芳香性。LCDR1中的残基Gln27、HCDR1中的Asn28、HCDR2中的Asn55和HCDR3中的Asp102形成了一个极性和亲水性口袋,以包含疏水核心。
《图3》

图3 HER2和HF作用复合物的3D结构。(a)根据抗体可变区框架的拓扑结构合理选择CDR残基。HF的CDR区结合环的俯视图显示为空间填充模型(i)和带状结构(ii),突出显示了重链CDR-H1(黄色)、CDR-H2(绿色)和CDR-H3(蓝色)以及轻链CDR-L1(粉色)、CDR-L2(洋红色)和CDR-L3(红色)中优化了的残基。本图和后续图中使用了Kabat编号。(b)HER2结合赫赛汀的3D结构[(i)HER2-赫赛汀复合物结构模型]和HER2结合HF的3D结构[(ii)HER2-HF复合物结构模型]。
HER2和新型抗体HF的3D复合结构如图3(b)所示。比较HER2和赫赛汀晶体结构,HF和HER2之间的结合模式比赫赛汀和HER2之间的结合模式更紧密。此外,除了由赫赛汀特异性识别的表位D1之外,新型抗体HF还识别了新表位D2。
《3.2. 新型抗体HF的构建与鉴定》
3.2. 新型抗体HF的构建与鉴定
使用逆向翻译去设计用于哺乳动物细胞表达的新型抗HER2抗体HF(中国专利ZL200910131355.3)的可变区序列。采用重叠PCR的方法去克隆HF VH和VL基因片段,将可变区插入含有IgG1恒定区的表达载体,构建抗体HF的表达载体。根据使用固定山羊抗人IgG的夹心酶联免疫吸附试验可知,HF抗体的表达水平为5~10 g·mL-1(数据未显示)。使用蛋白A层析亲和纯化抗体,通过考马斯亮蓝染色的十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(非还原型)显示抗体分子量(MW)约为150 kDa的单一条带,与预期吻合。电泳分析(还原型)显示两条条带,与单个重链(50 kDa)和轻链(25 kDa)的预期MW一致(数据未显示)。N端测序和基质辅助激光解吸电离飞行时间肽质量指纹鉴定给出了轻链和重链的预期序列(中国专利ZL200910131355.3)。
通过流式细胞仪分析评估HF的抗原识别能力。将几种人肿瘤细胞系与抗体共同孵育。结果表明,HF只能与HER2过表达细胞系(如SKOV3、SKBR3、MCF7和T47D)结合[图4(a)]。为了进一步确定HF结合HER2的特异性,我们将编码HER2细胞外结构域(ECD)的基因融合,并在293T细胞膜上表达,以增强型绿色荧光蛋白(EGFP)转染细胞作为阴性细胞对照。流式细胞术[图4(b)]和荧光显微镜检测[图4(c)]结果均表明,过表达HER2的293T细胞可以被特异性识别,而只表达EGFP的细胞不能被特异性识别。数据支持HF特异识别HER2的观点。
《图4》
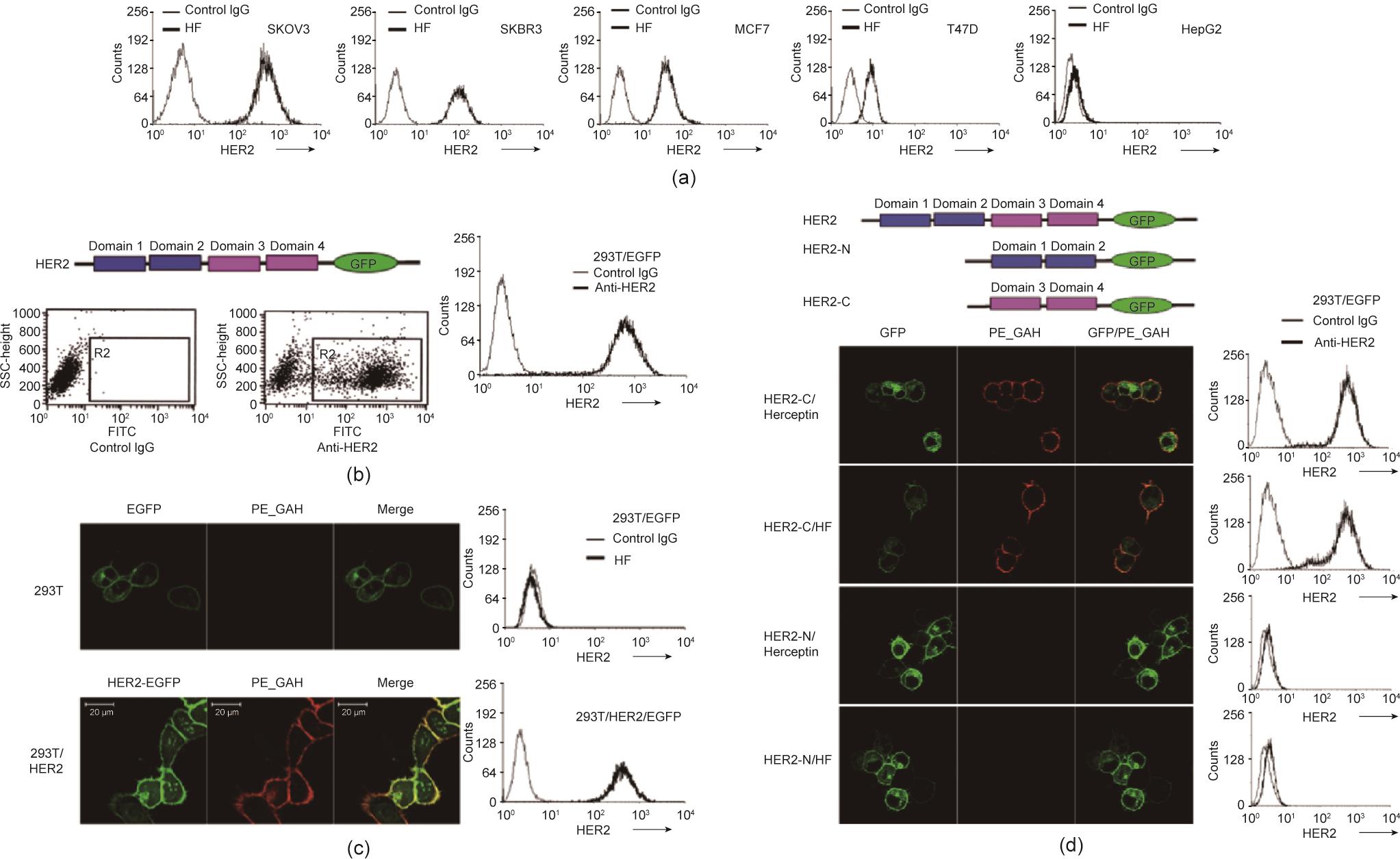
图4 HF在体外与膜HER2抗原结合。(a)HF与HER2阳性细胞系上的膜抗原结合;通过流式细胞术分析(b)和荧光显微镜检测(c)可见,HF与HER2转染的293T细胞膜抗原结合;(d)赫赛汀和HF均与HER2 C端结构域结合,但不与N端结合。SSC:侧面散射;GFP:绿色荧光蛋白;FITC:异硫氰酸荧光素;R2:选择区域2细胞进行流式细胞术分析;EGFP:增强型绿色荧光蛋白;HER2-C:HER2 C端结构域;HER2-N:HER2 N端结构域。
HER2 ECD包含两个结构域:N端结构域和C端结构域。这些结构域介导不同的生物学功能。当设计HF抗体时,表位理论上位于C端。为了验证设计的合理性并确定HF的表位,我们构建了两个截短突变体HER2-C和HER2-N。荧光显微镜和流式细胞术(FACS)试验的数据表明,与赫赛汀相似,HF与HER2 C端结构域结合,这与理论设计的初衷一致[图4(d)]。
《3.3. HF的表位重叠,但与赫赛汀不同》
3.3. HF的表位重叠,但与赫赛汀不同
基于X射线晶体数据和HER2的分子模型预测3个潜在表位:550/552/553、560/561/562/565和583/585。HER2与赫赛汀形成的复合物的X射线晶体数据显示,赫赛汀表位为560/561/562/565,而基于HER2/HF复合物的理论结构可见,HF识别的是由560/561/562/565和583/585形成的空间表位。为了证实这些预测,我们通过丙氨酸置换构建了一系列膜HER2突变体(均被丙氨酸替代),命名为:mHER2,包括550/552/553、560/561/562/565和583/585突变体;mHER2-M1,550/552/553突变体;mHER2-M2,560/561/562/565突变体;mHER2-M3,583/585突变体[图5(a)]。这些突变体在与EGFP融合后,表达在293T细胞膜表面。用赫赛汀或HF孵育后再用PE_GAH孵育,通过FACS和荧光显微镜检测分析细胞。结果表明:① mHER2不能被赫赛汀或HF识别[图5(b)、(c)];②表位550/552/553不参与HER2与赫赛汀或HF之间的相互作用,而突变的mHER2-M2不被赫赛汀或HF识别,表明560/561/562/565是HF和赫赛汀共同识别的表位[图5(d)];③ mHER2-M3与赫赛汀相互作用,但不与HF相互作用。总之,这些发现表明HF的表位是由560/561/562/565和583/585构建的空间构象,其与赫赛汀不同。
《图5》
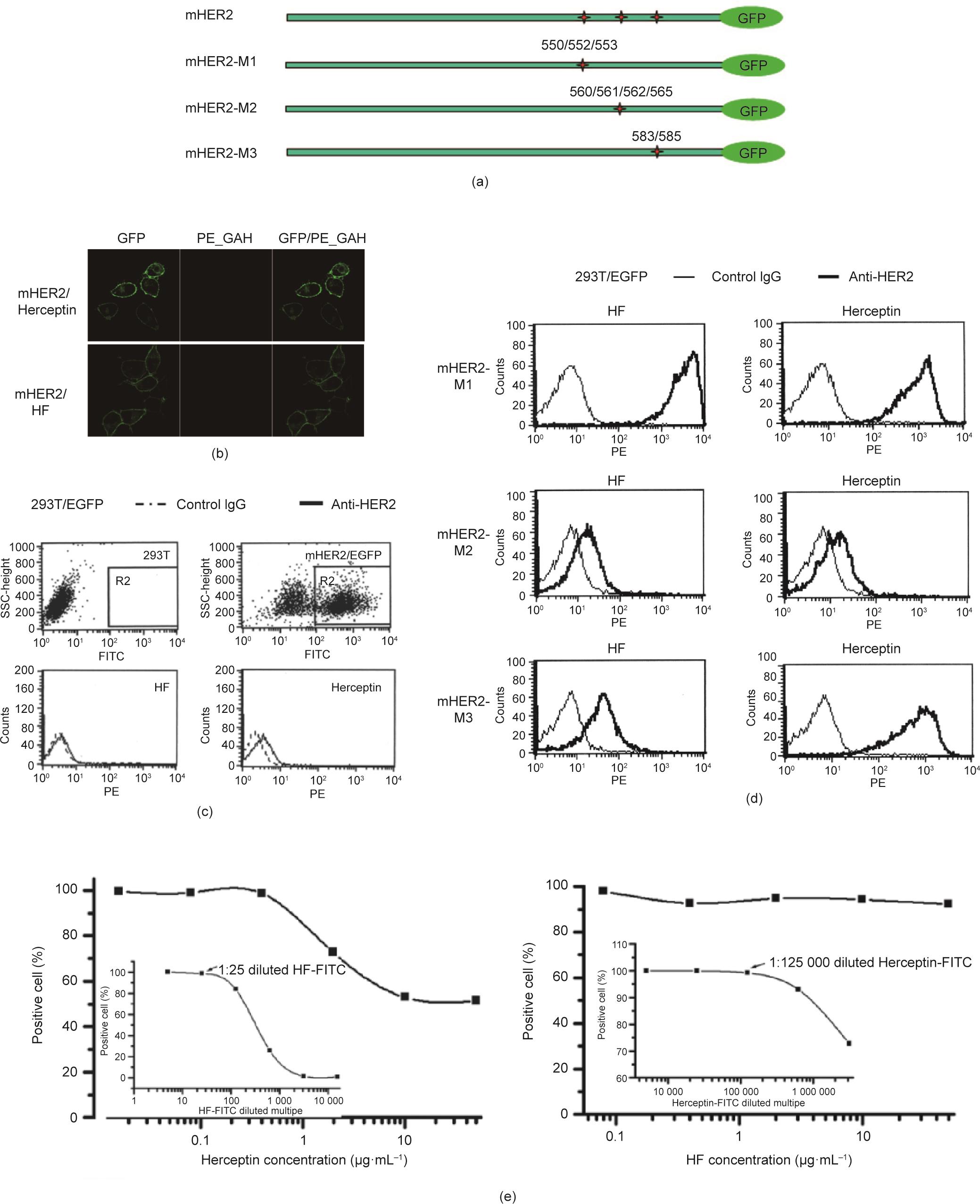
图5 HF的表位与赫赛汀不同。(a)设计的三种HER2突变体,即mHER2-M1~M3,其位点550/552/553、560/561/562/565或583/585被丙氨酸取代。mHER2是三重突变体。荧光显微镜(b)和流式细胞术检测(c)显示,HF和赫赛汀没有/具有非常弱的结合mHER2的能力。(d)HER2 583/585位点在HF结合中似乎比赫赛汀更重要,因为mHER2-M3/HF结合的平均值弱于mHER2-M3/赫赛汀。(e)HF的表位似乎与赫赛汀重叠,但与赫赛汀的表位不完全相同,因为赫赛汀可以抑制HF与HER2的结合,最大抑制率约为50%;但是50 μg·mL-1 HF不能抑制赫赛汀与HER2的结合。嵌套图显示了每个分析中使用的FITC标记抗体(1∶25稀释HF-FITC或1∶125 000稀释赫赛汀FITC)的实验浓度。
此外,通过流式细胞术分析了两种抗体的竞争效应。标记抗体与SKOV3细胞孵育,而另一种抗体(裸体)作为竞争剂掺入。赫赛汀能抑制HF与HER2的结合,当赫赛汀浓度达到10 g·mL-1时,最大抑制率在50%左右。相反,HF不能抑制赫赛汀结合活性。这种竞争概况[图5(e)]也表明HF的表位与赫赛汀的表位略有不同。
《3.4. HF的体外抗肿瘤活性》
3.4. HF的体外抗肿瘤活性
用稀释的HF孵育HER2阳性肿瘤细胞,测定其体外抗细胞增殖活性。设定赫赛汀和IgG作为对照。HF轻度抑制HER2阳性细胞增殖。在浓度大于或等于4 g·mL-1时,约10%的SKOV3细胞增殖受到抑制[图6(a)]。在HER2low + MCF7细胞中,几乎没有观察到抗增殖活性[图6(b)],这表明HF直接抗肿瘤活性不明显。
《图6》

图6 HF体外抗肿瘤活性。HF对HER2阳性SKOV3(a)或MCF7细胞(b)的增殖表现出弱抑制/无抑制;(c)HF有效介导HER2阳性SKOV3、SKBR3和MCF7癌细胞的ADCC功能;(d)HF的亲和力高于赫赛汀,主要原因在于HF具有更高的Kon和更低的Koff常数。ADCC:抗体依赖细胞毒性;Kon:结合速率常数;Koff:解离速率常数。
然而,使用人PBMC作为效应细胞,HF可在表达HER2的SKOV3、SKBR3和MCF7癌细胞中有效介导抗体依赖性细胞毒性(ADCC)[图6(c)]。此外,杀伤活性与细胞表面的HER2表达水平成正比。在最大检测浓度(20 g·mL-1)下,抗HER2抗体可杀死50%~60%的SKOV3细胞、30%~40%的SKBR3细胞和20%~30%的MCF7细胞,HER2表达顺序为SKOV3 > SKBR3 > MCF7(见附录A中的图S1)。有趣的是,尽管HF和赫赛汀的最大杀伤率相似,但在较低浓度下,HF显示出比赫赛汀更有效的杀伤活性。在10 g·mL-1浓度下,HF杀死30.24% ±5.12%的SKOV3细胞,而赫赛汀杀死13.73% ± 1.94%的细胞。在SKBR3和MCF7细胞中也观察到类似的结果。除表位差异外,HF可能具有更高的亲和力,因为更高的亲和力是影响ADCC活性的关键因素之一[16]。通过Fortebio法测量这些抗HER2抗体的相对亲和力[17],结果表明,HF的结合速率常数(Kon)高于赫赛汀,而HF的解离速率常数(Koff)低于赫赛汀,这意味着HF的相对亲和力高于赫赛汀[图6(d)]。
《3.5. HF在异种移植小鼠模型中的体内活性》
3.5. HF在异种移植小鼠模型中的体内活性
为了评估新型抗体HF是否在体内抑制肿瘤的增殖,我们通过给免疫受损小鼠接种HER2过度表达的癌细胞来建立实体瘤模型。
在BALB/c裸鼠腋下接种SKOV3细胞,8 d后开始HF给药(静脉注射,首剂5 mg·kg-1,然后每周一次2.5 mg·kg-1,持续4周)。每三天用卡尺测量一次肿瘤,用公式计算每个肿瘤的体积:体积= (
《图7》
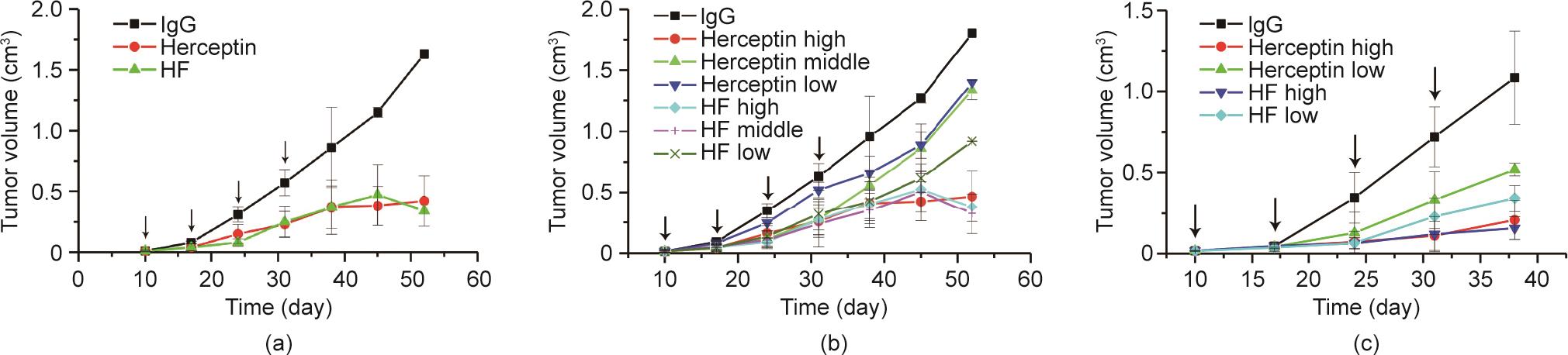
图7 HF体内抗肿瘤活性。(a)、(b)在SKOV3异种移植小鼠模型中,HF明显抑制肿瘤生长,且呈剂量依赖性。在中、低剂量时,HF明显比赫赛汀表现出更好的作用。(c)HF抑制BT474异种移植小鼠模型中的肿瘤生长。HF也具有比赫赛汀更好的抑制能力,尤其在低剂量组中,最大抑制率可达80%。
为更好地确定抗HER2抗体在治疗过程中是否存在剂量依赖性,我们使用较低剂量的抗体进行了第二次SKOV3异种移植小鼠实验。本实验给予抗HER2抗体,首次剂量为1.25 mg·kg-1、2.5 mg·kg-1或5 mg·kg-1(静脉注射),随后剂量减半,每周1次,连续4周。在HF给药和赫赛汀给药小鼠中均观察到显著的剂量依赖性抗肿瘤活性[图7(b)]。值得注意的是,尽管在5 mg·kg-1时没有显著差异,但HF在较低剂量下显示出了比赫赛汀更有效的抗肿瘤活性。与用对照抗体处理的小鼠相比,1.25 mg·kg-1、2.5 mg·kg-1和5 mg·kg-1 HF给药小鼠在52天时表现出的肿瘤生长平均抑制率分别为49.1% ± 1.6%、80.8% ± 9.1%和78.9% ± 2.4%。而使用赫赛汀时,抑制率分别为22.4% ± 1.9%、25.8% ± 4.5%和74.2% ± 11.5%。在接种另一种人乳腺癌细胞系BT474的BALB/c裸鼠的实体瘤模型中也获得了相似的结果[图7(c)]。
《3.6. HF积累的体内观察》
3.6. HF积累的体内观察
在SKOV3异种移植BALB/c的裸鼠中观察HF抗体的体内分布。在肿瘤细胞皮下接种后的第28天,用Cy5.5标记的HF抗体以25 mg·kg-1的剂量静脉给药。可见HF逐渐渗透到肿瘤中并聚集在肿瘤部位,然后在4 h内扩散到整个肿瘤区域,注射后48 h,HF分子仍存在于肿瘤中[图8(a)]。处死小鼠,对肿瘤和器官进行尸检,发现HF抗体在SKOV3诱导的肿瘤中选择性蓄积。除肝脏和肾脏中的轻微蓄积[图8(b)]外,宿主重要器官中无信号,这可能是由于代谢和排泄所致。为了进一步确定HF在体内的结合特异性,我们分别在异种移植HER2阳性SKOV3肿瘤和HER2阴性HepG2肿瘤的裸鼠中重复该实验。结果发现,仅SKOV3异种移植物可蓄积HF,而在HepG2异种移植物中未观察到相似的蓄积[图8(c)]。上述数据有力地表明,静脉注射后HF可以选择性地在HER2+实体瘤中蓄积。
《图8》
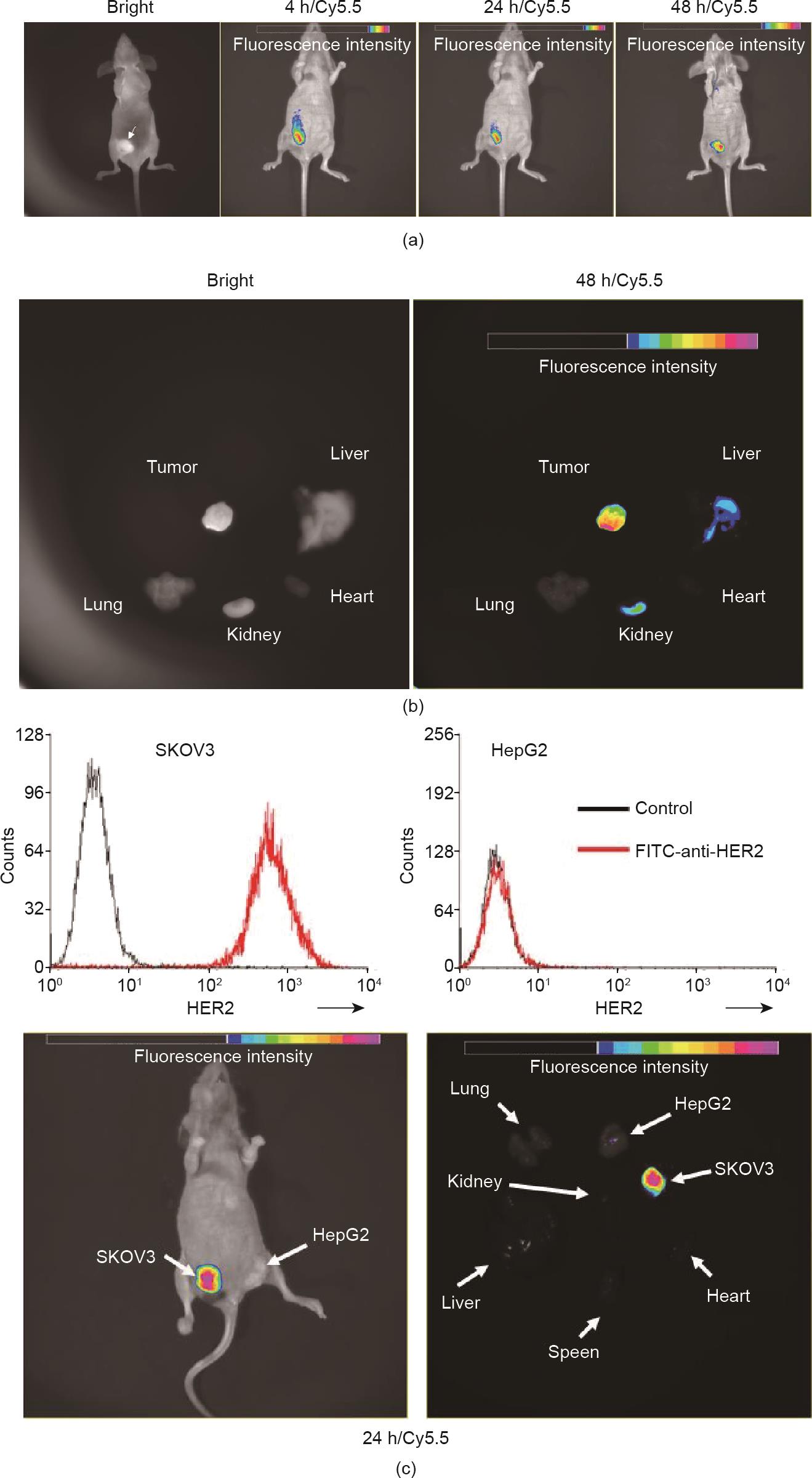
图8 HF的体内靶向和积累。(a)Cy5.5-HF主要以剂量依赖性的方式在肿瘤中积聚;(b)HF主要见于肿瘤,然而由于代谢和排泄,一些HF也可见于肝脏或肾脏;(c)HF在24 h后没有靶向HER2阴性的HepG2异种移植物。
《4. 讨论》
4. 讨论
在过去30年中,各种技术进步极大地促进和发展了单克隆抗体治疗。尽管首先发现了鼠单克隆抗体,但因免疫原性等副作用,单克隆抗体未能成为临床治疗的主要选择。嵌合抗体虽展现出了较低的免疫原性,但可变区仍含有小鼠序列。人源化单克隆抗体实现了小鼠序列的进一步减少,随后是全人治疗性单克隆抗体。目前,全人抗体在感染、恶性肿瘤和炎症等疾病的临床治疗中显示了广泛的用途。
目前,众多技术可以用于制备全人抗体,包括噬菌体文库技术、转基因小鼠技术、B细胞永生化、人-人杂交瘤、单细胞PCR等。然而,大多数全人单克隆抗体疗法,无论是在用的还是在开发中的,其抗体均来自非人种属(因为当前技术尚不可能永生化和克隆人B淋巴细胞)。目前的方法包括从丝状噬菌体/酵母中获得人类基因编码的单链抗体片段(scFv),或从染色体携带人类免疫球蛋白基因的转基因小鼠中筛选抗体。
计算机技术如今在药物设计领域受到重点关注。基于结构的小分子设计技术近年来已成为药物发现过程中,特别是许多高活性分子的发现过程中的重要组成部分[18]。由于大蛋白在处理过程中的计算复杂性和工程蛋白的信息稀缺,蛋白的计算设计具有很大挑战性[19-20]。最近,蛋白质设计领域又取得了许多新成就,包括重新设计具有内部结构域-结构域界面的内切核酸酶[21]、设计蛋白质中的新折叠[22]、设计周质结合蛋白中的特殊酶活性[23],以及DNA酶抑制剂的突变[24]等。用于抗体分子设计的计算机技术已被证实可有效提高抗体的药物特性,如亲和力[3,25]、效应功能[5]和稳定性[6-7]。然而,计算机辅助设计技术在新生抗体设计领域的应用仍然相对较少[26]。
本研究提出了一种利用计算机辅助虚拟筛选和抗体结构信息生成方法设计新型全人抗体的策略。此外,我们对新型抗体HF进行了鉴定,并将其与治疗性抗体赫赛汀进行了比较。
我们提出的策略原则上适用于任何基于功能表位空间构象设计的新生抗体分子。与当前技术相比,本方法具有如下几个优势:首先,它具备较强的亲和力且抗体是全人的,选择的抗体框架也来自于所收集的人抗体框架序列;此外,在新生抗体设计程序中筛选了最适合的关键残基(图2),使得HF的人源化水平高于赫赛汀。同时,HF的结合能力也优于赫赛汀(图6)。其次,抗原-抗体复合物结构可以可靠地用于新生抗体设计的参考。虽然计算机辅助蛋白质虚拟对接技术正在迅速发展,但当以理论结构为起点时,该技术仍存在许多局限性。然而,抗体结构相当保守,抗原和抗体的结合域均位于CDR区和抗体识别的抗原表位区,该信息有助于更准确地预测抗原-抗体复合物模型。借助位点诱变等其他实验数据,可以进一步优化抗原-抗体对接构象。
总而言之,用于癌症、免疫相关疾病、传染病等疾病的有效治疗的抗体药物的发现,客观上需要新的突破性技术的出现。本方法即是独立于常用的杂交瘤技术、抗体人源化和亲和力成熟等技术之外的有效的抗体发现技术,从而为基础研究、成像和临床应用设计新型全人抗体。











 京公网安备 11010502051620号
京公网安备 11010502051620号




