
《1. 引言》
1. 引言
氯酚(CP)是一大类在化工和制药行业中释放到环境中的重要污染物,也是聚合物、染料、纺织品、树脂和炸药生产制造过程中产生的副产物。CP及其衍生物对人类和其他生物具有致突变性、致癌性和免疫原性,其处理、处置和管理已成为企业参与者和卫生部门面临的严峻挑战。大多数类型的CP难以被微生物降解,因此必须开发更有效的方法减少其对水环境和生态系统的污染。将清洁电子作为反应物驱动的电化学氧化由于具有高效性、清洁性、过程集成化和自动化等优势,在分散式水处理领域备受关注[1‒3]。电化学氧化通过直接电子转移(DET)氧化和电解水产生·OH介导的间接氧化降解CP污染物。
由于低浓度CP的氧化速度快于其向阳极表面的传质速度,因此CP的电化学氧化通常受到扩散限制。电活性膜(REM)通过穿流式电解操作,为有效提高传质效率提供了一种可靠方法[4‒5]。与机械搅拌和旁流式电解相比,REM可在穿流式电解中借助对流强化传质速率[6]。因此,膜电极在污染物去除、自清洁膜、资源回收、海水淡化等净水领域具有广阔的应用前景[7‒9]。
目前REM的绝大多数研究处在精准控制的实验室阶段,因此在基础研究和工程化应用之间仍存在差距。将反应器从实验室规模放大到工程应用的过程通常要经历“死亡之谷”,然而缺乏相应准则难以保证膜电极放大成功。不同于传统化学和生化反应器依赖几何学、动力学和热力学特性的相似性放大准则,电极构型在REM放大过程中扮演着至关重要的作用[2,10‒11]。大多数穿流式电化学反应器采用推流式过滤构型[12‒13],这种配置由多孔导电材料如碳纳米管(CNT)或Ti4O7 [14‒15]制成的两块平行平板电极(PPE)构成。尽管这种电极配置为电极材料的性能测试提供了一个简单且易于操作的平台,但在电化学反应器规模放大上仍存在挑战,主要原因为增加电极间距或反应器体积导致欧姆电阻增加,同时电极面积与反应器体积之比降低。因此迫切需要开发一种高效、可靠且易于放大的电极构型,以期实现面向精准化工程应用的反应器设计。
合理的电极构型原则上应:①具备合理的电场分布和较低的欧姆电阻;②能够有效去除氯酚类污染物;③能够组件化,便于工程放大和实际应用。研究表明圆柱形的管状电极在固体氧化物燃料电池(SOFC)、聚合物电解质膜燃料电池(PEMFC)和钒氧化还原液流电池(VRFB)[13,16‒18]等能量转化体系中更有利于提高电流密度和产电能力。受这些成功应用的启发,本研究试图开发一种同轴管式电极(TCE)构型来跨越REM规模放大的“死亡之谷”,以便实现电化学去除CP污染物的工程应用。
本研究开发了一种由内部亚氧化钛管(TiSO)(陶瓷膜阳极)和外部不锈钢(SS)管(阴极)组成的TCE构型,能够实现废水在电极管之间从阳极流向阴极(AC)或从阴极流向阳极(CA)。其中管状TiSO阳极材料具有与金属和陶瓷相媲美的导电性和耐腐蚀性。由于其高导电性、良好稳定性、高析氧电位和低成本等独特优势,在水处理领域中拥有广阔前景[19]。首先,利用软件模拟和电化学试验考察了不同电极构型下的电场分布;其次,测试了典型难降解氯酚类污染物(即2,4-DCP)在不同穿流方向即AC模式和CA模式下的电化学去除效能,结合理论计算和实验数据阐明了相关机理;最后,提出了一种增加TCE组件数量的策略用于放大穿流式反应器,并验证了其在单程穿流模式下工程化应用的适用性。本研究的主要目标是确定面向用于REM反应器放大的合理电极构型。
《2. 材料与方法》
2. 材料与方法
《2.1. 材料与试剂》
2.1. 材料与试剂
所有采购的试剂如无特殊声明均为分析纯,并且在使用时未进一步纯化。高氯酸钠(NaClO4)、2,4-二氯苯酚(2,4-DCP)和色谱纯级甲醇购自国药集团化学试剂有限公司,溶液由Milli-Q系统[18.2 MΩ·cm,(22 ± 1) ℃]获得的超纯水配制。TiSO电极根据先前研究中的方法进行设计和制备[19]。
《2.2. 实验装置与操作》
2.2. 实验装置与操作
电解实验在室温[(25 ± 1) ℃]下进行,有机玻璃圆柱电解池的总体积为3.8 L(220 mm × Φ150 mm)。电解池包含管状的TiSO阳极(外径为30 mm,厚3 mm,长143 mm,几何面积为132.9 cm2)和SS阴极(几何面积为140 cm2),电极通过铜线连接到直流电源上。试验前,将TiSO电极在0.5 mol·L-1 NaClO4溶液中以30 mA·cm-2的电流密度活化10 min。初始进水为0.1 mol·L-1 NaClO4和10 mg·L-1 2,4-DCP,pH中性(7.2 ± 0.2),电解池以不再循环的单程穿流模式运行。实验在不施加电流的条件下先稳定10 min,再以特定的时间间隔从出水中采集5 mL样品,用醋酸纤维素过滤器(0.22 μm)立即过滤并进行化学分析。
体系的水力停留时间(HRT, s)由下式计算:
(1)
式中,J为膜通量(m·s-1);d为TiSO阳极的厚度(m);A为阳极面积(m2);Q为流速(m3·s-1)。
《2.3. 电化学表征》
2.3. 电化学表征
以SS和Ag/AgCl [内置饱和KCl溶液,相对标准氢电极(SHE)为0.198 V]分别为对电极和参比电极进行电化学测试。采用三电极体系对2,4-DCP以100 mV·s-1的扫描速率进行循环伏安(CV)测试。电化学阻抗谱(EIS)测试施加的正弦交流扰动信号频率为10~100 kHz,振幅为5 mV。
《2.4. 分析方法》
2.4. 分析方法
采用配备光电二极管阵列检测器(PDA)和反相C18色谱柱(50 mm × 2.1 mm, Waters,美国)的超高效液相色谱(UPLC)测定2,4-DCP浓度,流动相由水/甲醇(30∶70, V/V)组成,流速为0.1 mL·min-1。采用UPLC联合电喷雾电离质谱(UPLC-ESI/MS, Agilent 6460,美国)对2,4-DCP降解中间产物通过质荷比进行定性分析,流动相为A(超纯水)和B(色谱纯乙腈)。在负扫描模式下以0.15 s的扫描时间采集了质荷比(m/z)在80~220范围内的高分辨率连续质谱,其中疑似中间产物在碰撞池中电离破碎。电离源设置为:毛细管电压3 kV,温度200 ℃,脱溶剂温度550 ℃,对20 μL样品重复分析以确保质谱结果的准确性和可重复性。化学需氧量(COD)采用LH-3BA分光光度计(中国联华科技有限公司,化学试剂型号:LH-D/E)分析。
去除单位量级污染物的标准化能耗(EEO)计算如下[20]:
(2)
式中,EEO为将有机污染物浓度降低一个量级所需的体积能耗(kW·h·m-3);U为平均池电压(V);j为电流密度(mA·cm-2);A为阳极面积(m2);Q为流量(m3·h-1);I为电流(A);Cf和Cp分别为进水和滤液中的污染物浓度(mg·L-1)。
电化学氧化的电流效率为:
(3)
式中,CODf和CODp分别为进水和滤液中的COD浓度(mg·L-1);b为电子转移数的化学计量系数;F为法拉第常量(96 485 C·mol-1);V为t时间内的处理体积(L);
《2.5. 电场模拟》
2.5. 电场模拟
采用COMSOL Multiphysics软件(5.5版本;Palo Alto,美国),根据电场强度定义对PPE、管状/平面电极(TPE)和TCE电极构型中的电场分布进行模拟:
(4)
式中,E为电场;
《2.6. 理论计算》
2.6. 理论计算
采用高斯软件进行密度泛函理论(DFT)计算。根据Marcus理论可计算出依赖电极电位的DET氧化反应的活化能(Ea, kJ·mol-1)。DET还原反应的计算类似。
(5)
式中,λf为电子转移氧化反应的重组能(kJ·mol-1),是具有相同优化结构的产物与反应物的ΔG0(kJ·mol-1)差值;E为相对SHE的电极电位(V),E0为反应物DET反应的标准电位(V),在热力学上由下式确定:
(6)
式中,ΔrG0为反应物氧化的自由能(kJ·mol-1),可由几何结构优化的反应物和产物的ΔG0确定。n(n = 1)为电子转移数;
《3. 结果与讨论》
3. 结果与讨论
《3.1. 不同电极构型的电场分布》
3.1. 不同电极构型的电场分布
电场分布与带电离子在电场中的传质转运密切相关,因此电场的空间分布显著影响电化学反应[13,22‒25]。事实上,电化学反应仅能发生在电极表面电场线到达的区域,反之亦然。由于电场分布与电极的构型配置密切相关,本研究采用COMSOL建模对废水处理中三种典型电极构型(PPE、TPE和TCE)[13]的静电场分布进行可视化和定性分析。如图1所示,几何对称的PPE和TCE电极构型展现出最均匀的电场分布,而TPE构型中静电场具有非对称性,与Hankin等[23]在光电化学反应器和Sun等[26]在电化学反应器中报道的结果相似。这一结果清楚地表明,采用PPE和TPE构型能够实现阳极面积的完全有效利用。然而鉴于等势线总是处处垂直于电场线,TPE构型中管状阳极面积仅有小于1/3呈现电化学活性。电场总是由阳极面指向阴极面,由于电荷数量相等且电性相反,电场在电极间区域叠加,而在电极之外的区域内抵消为零[26]。因此,只有垂直于电场线的电极表面才具备电化学活性,而其他区域不会发生电化学反应。SS圆盘上的电沉积结果可视化地验证了这一结论(附录A中的图S1)。其中仅在电极正面发现了电沉积的Cu膜,背面边缘少量Cu膜可能是由Cu2+在浓度梯度下的扩散所致。
《图1》
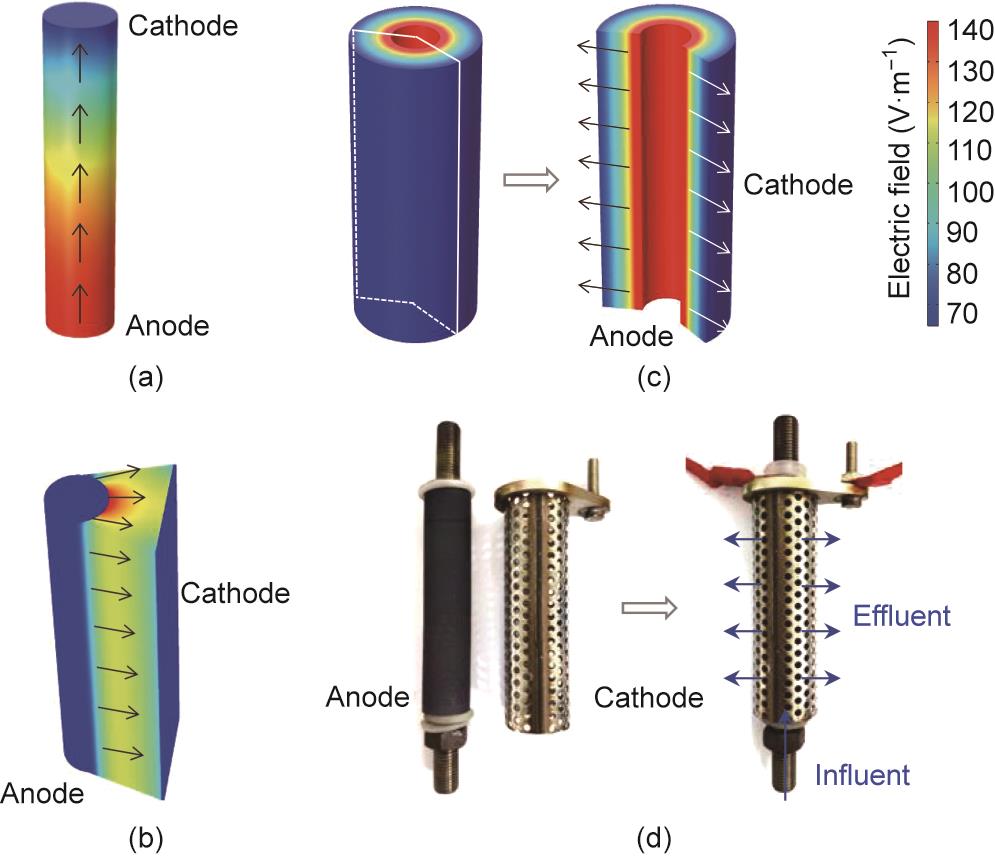
图1 COMSOL模拟不同电极构型的静电场分布。PPE(a)、TPE(b)和TCE(c)的主视图和剖面图;(d)TCE组件的实物图。
对于同一阳极,阴极几何形状对电场分布有显著影响。由于离子沿着电场线扩散,显然电极间均匀的电场分布对有效利用电化学活性区域至关重要,已在电化学高效消毒中得到证实[24]。这些结果表明在设计穿流式电解池时,电极表面应处处与电场线正交,以便最大限度地利用电极的有效电化学活性面积。根据这一原理,PPE设计更适用于推流式电解,而TCE设计更适用于穿流式电解。
《3.2. 不同电极构型的欧姆电阻》
3.2. 不同电极构型的欧姆电阻
当电流通过电解池发生阳极极化时,电解池上的池电压U(V)可由平衡电位与过电位共同决定[27]:
(7)
式中,Umin为电解所需的最小理论电位(V);ηA为取决于催化剂种类的活化过电位(V);ηD为浓差极化引起的扩散过电位(V);而ηohm为由电极材料、构型和电解质引起的欧姆过电位(V)[27]。在给定的穿流式电解体系下,ηohm是电解池电压的主要贡献者,决定了恒流电解下的能量消耗。根据欧姆定律可计算欧姆过电位:
(8)
式中,ΣRohm为欧姆电阻的总和(Ω);I为电流(A)。
值得注意的是,电极构型能够显著影响欧姆电阻的大小。例如,在相同的阳极直径(29 mm)、轴向长度(142 mm)和电解质(0.1 mol·L-1 NaClO4, pH 6.5)条件下,根据EIS谱图数据中的x轴截距,TCE构型的ΣRohm值(2.1 Ω)分别较PPE(19.8 Ω)和TPE(12.6 Ω)构型低89.3%和83.3% [图2(a)]。这种差异可能是电极间的电解质在各自电场作用下的离子转运差异所致。其中,PPE构型的欧姆电阻与反应器的轴向长度成正比,如下所示:
(9)
式中,κ为电解质的比电导率(S·m-1);L为管式反应器的轴向长度(m);A为电极面积(m2);r为电极半径(m)。根据方程(9),
(10)
式中,r1和r2分别为管式反应器阳极和阴极的半径。
《图2》

图2 (a)不同电极构型和不同轴向长度的PPE(b)和TCE(c)管式电解池反应器的EIS谱图;(d)TCE构型中总内阻(ΣRohm)与轴向长度的关系。
尽管在实验室规模的小型电解池中,PPE和TCE构型的ΣRohm值仅存在细微差异,但随反应器体积的增加,两种构型的ΣRohm值呈现截然相反的特征[图2(b)、(c)]。其中,欧姆电阻在TCE体系放大过程中不断降低,这一特点对低电导率水质(如生活污水和地下水)[28]尤为重要。如图2(d)所示,当管式反应器轴向长度从1 cm增加到14 cm时,PPE构型的ΣRohm值能够达到TCE构型的16倍[方程(9)和方程(10)]。这一结果表明结构紧凑的TCE构型由于电极间距小,电化学活性面积大,显著增强了电子转移能力并降低了欧姆内阻[29]。
实验结果结合理论验证表明,TCE构型具有均匀的电场和较低的欧姆电阻,更有利于REM的工程放大。类似地,Lei等[30]在柱状电化学反应器中采用可放大的管式SS阴极,对磷酸盐实现了连续173 d的高效回收(> 50%)。这种TCE构型的独特优势使其在膜电极体系放大中得以工程应用[31]。接下来,本研究将聚焦到TCE体系在废水处理领域中的工程应用潜力。
《3.3. REM组件去除2,4-DCP的性能》
3.3. REM组件去除2,4-DCP的性能
在TCE构型中电极间电场分布均匀,表明能够有效发生法拉第反应。以阳极分别作上游(AC模式)和下游(CA模式)的流动顺序对电化学反应产生重要影响。为此,本研究采用TiSO阳极/SS阴极构成同轴管式REM反应器,考察在不同流动方向、电流密度和流动速度下2,4-DCP(10 mg·L-1)在NaClO4(0.1 mol·L-1, pH 6.5)电解质中的电化学去除效能。2,4-DCP通过在阴极(SS)和阳极(TiSO)分别接受和失去电子而发生电催化还原和氧化反应。其中,通过电极间的水流传质实现阳极氧化耦合阴极还原反应。
在电流密度5~30 mA·cm-2和流速15~120 mL·min-1下进行系列实验,考察了电化学去除2,4-DCP。如图3(a)所示,AC模式下电流密度从5 mA·cm-2增加到30 mA·cm-2,2,4-DCP去除率从5%~28%增加到18%~71%。由于阳极氧化10 mg·L-1 2,4-DCP的极限电流密度为0.6 mA·cm-2,远低于施加的电流密度,因此2,4-DCP降解受到扩散控制。尽管如此,当流速在15~120 mL·min-1范围内增加时,传质系数从2.5 × 10-6 m·s-1增至2 × 10-5 m·s-1。反应器连续运行时保留时间从160 s降至20 s,导致AC模式下2,4-DCP去除率降低70.7%~78.5%。Wang等[32]同样也报道了在较低流速下的电化学氧化效率高于较高电流密度。类似地,在CA模式下2,4-DCP去除率随电流密度的增加从30%~85%(5 mA·cm-2)增加到81%~99%(30 mA·cm-2),并随流速的增加从85%~99%(15 mL·min-1)降至30%~81%(120 mL·min-1)。CA模式下的表观速率常数较AC模式高一个数量级[图3(c)和(d)],表明阴极/阳极的协同作用对流动方向具有内在依赖性。附录A中的图S2比较了在电流密度为20 mA·cm-2且流速为60 mL·min-1时,两种操作模式下2,4-DCP的去除效果。2,4-DCP在CA模式下电解5 min后几乎完全去除,而在AC模式下仅去除16.8%。流速为60 mL·min-1时,将电流密度从5 mA·cm-2提高到30 mA·cm-2,CA模式下的表观速率常数从0.19 min-1增加到0.21 min-1,为AC模式下的11.2~78.1倍[图3(c)]。电流密度为20 mA·cm-2时,将流速从15 mL·min-1增加到120 mL·min-1,伴随穿流式电解改善了多孔阳极结构中的对流扩散,使得氧化速率得到显著提升。因此,CA模式下的表观速率常数从0.13 min-1增加到0.28 min-1,大约是AC模式下的6.7~11.2倍[图3(d)]。可见2,4-DCP的氧化动力学在CA模式下对流速更为敏感,面积归一化的表观速率常数(0.036~0.079 m·s-1)较AC模式高近一个数量级(附录A中的表S1)。因此废水从阴极流向阳极时,阴极活化(下文讨论)后的传质步骤主导·OH介导的间接氧化反应。
《图3》
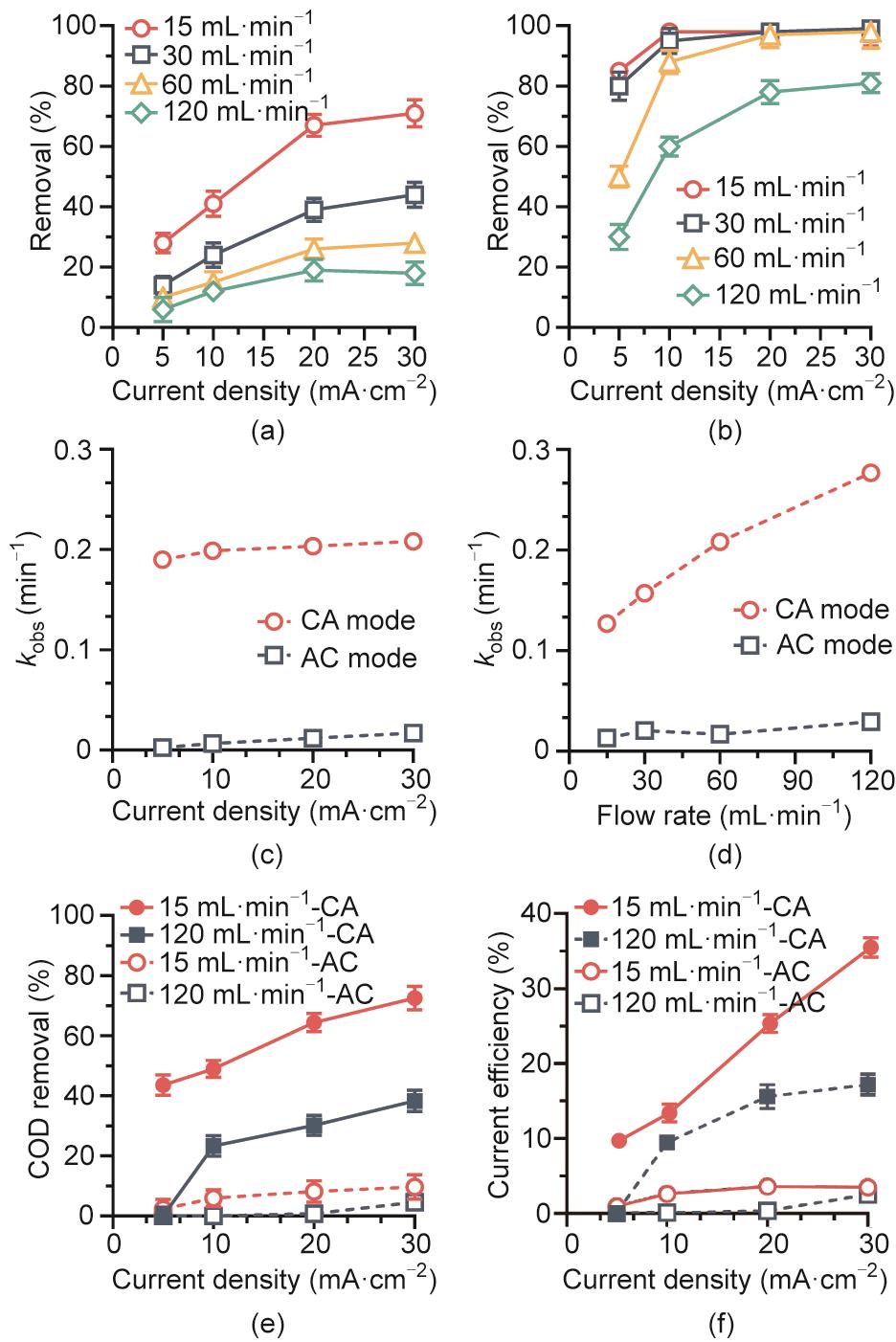
图3 TCE反应器中2,4-DCP的去除效能与流向、电流密度和流速的关系。AC(a)和CA(b)模式下去除率与电流密度和流速的关系;(c)流速15 mL·min-1时去除动力学与电流密度的关系;(d)电流密度20 mA·cm-2时去除动力学与流速的关系;两种操作模式下COD去除率(e)和电流效率(f)与电流密度和流速的关系。AC:从阳极流向阴极;CA:从阴极流向阳极;进水溶液:[2,4-DCP] = 10 mg·L-1,电解液[NaClO4] = 100 mmol·L-1。
此外,电解水导致阳极和阴极附近产生H+和OH-,尤其在高电流密度下伴随流向差异,H+和OH-在AC模式和CA模式之间也存在传质差异。在CA模式下,阴极析氢反应(HER)产生的OH-扩散到阳极,能够中和阳极水氧化产生的质子,导致阳极附近的pH值略有上升(附录A中的图S3)。此时,2,4-DCP(pKa = 7.8)倾向于去质子化并通过静电作用吸附在阳极上,利于随后的阳极氧化。如附录A中的图S4所示,CV曲线表明2,4-DCP发生电氧化的最小电位为1.66 V vs SHE,并且氧化峰的强度对pH响应敏感且呈正相关。这表明在高pH条件下能促进2,4-DCP在阳极氧化。2,4-DCP的典型中间产物,如2-CP和4-CP的pKa值为8~9 [33],同样也使其在高pH值下更具反应活性。这些因素使TCE反应器能够在CA操作下降解2,4-DCP,COD的去除率和电流效率最高可分别达到72.5% [图3(e)]和35.5% [图3(f)],大幅高于AC电解模式下的去除效果。
《3.4. 流向对2,4-DCP降解的影响》
3.4. 流向对2,4-DCP降解的影响
为了进一步阐明基于TiSO阳极的TCE反应器对2,4-DCP的降解机制,采用UPLC-四极杆-飞行时间质谱(UPLC-Q-TOF),通过质荷比(m/z)分析解析了中间产物。在AC模式下,检测到2,4-DCP电化学氧化过程中的6种典型中间产物(附录A中的图S5)与先前的报道结果吻合[34‒35]。然而在CA模式下检测到不同的中间产物,表明流向改变了电催化反应顺序,从而决定了反应路径和选择性。附录A中的图S6显示了AC模式下主要中间产物如氯代二羟基苯(2或3;m/z = 142.9342)和氯代苯醌(4或5;m/z = 140.9376)的累积情况。AC模式涉及的反应主要包括2,4-DCP的直接和间接阳极氧化,以及氯代苯醌产物的直接阴极还原。相比之下,CA模式下观察到的特征中间产物为苯酚(m/z = 93.1581)和CP(8或9;m/z = 126.7408),其相对丰度在前2 min内增加,随后迅速下降,没有明显的积累。因此,推测CA模式下的相关反应为2,4-DCP的阴极还原和苯酚、对苯二酚(11)或邻苯二酚(12)的阳极氧化。
为了对流动模式的影响提供机理解释,采用DFT理论预测了两种模式下的自由能差异(图4)。如附录A中的图S7所示,电还原的活化能低于电氧化的活化能。具体而言,阳极氧化2,4-DCP、苯酚(10)、对苯二酚(11)和邻苯二酚(12)所需的最小电位分别为1.9 V vs SHE、1.85 V vs SHE、1.5 V vs SHE和1.4 V vs SHE。阴极还原CP(8或9)和氯代苯醌(4或5)所需施加的最高电位分别为-1.55 V vs SHE、-1.55 V vs SHE、-0.55 V vs SHE和-0.75 V vs SHE。考虑到二羟基苯酚(11和12)或苯醌(6和7)具有相似的氧化还原性质,因此本研究基于1,4-对苯二醌(7)简要分析了整体反应的自由能变化情况。根据附录A中的表S2可以确定,在阳极和阴极电位分别为2.38 V vs SHE和-1.45 V vs SHE时,AC和CA模式下的自由能分别为-488.4 kJ·mol-1和-684.7 kJ·mol-1,证实CA模式提供了热力学上促进2,4-DCP降解的反应路径。如图4(c)所示,AC模式下降解反应在热力学上受限于邻氯对苯二酚(3)向邻氯对苯醌(5)的转化,然而在阳极氧化前通过阴极还原能够有效缓解这一限制步骤。由于CP(8或9)脱氯所需的自由能低至-250.4 kJ·mol-1,并且通过施加较低的阴极电位(E0 = -1.15 V vs SHE)就能够在热力学上实现阴极还原。电催化还原产生的关键中间产物苯酚,将大幅增加后续在阳极界面通过DET氧化(图S7)和·OH攻击的反应活性,这合理解释了CA模式下的高反应活性和高选择性。与AC模式相比,阴极还原大幅降低了2,4-DCP的最小理论电位[方程(7)中的Umin],因此能够在降低电能输入的同时提高电流效率和电化学矿化效果(图3)。此外,CA模式的另一个优点是脱氯中间产物的生物毒性较低(附录A中的图S8)。通过这种方式能够有效地去除多种氯酚类污染物,平均去除率高于85%(附录A中的图S9;20 mA·cm-2和60 mL·min-1)。
《图4》
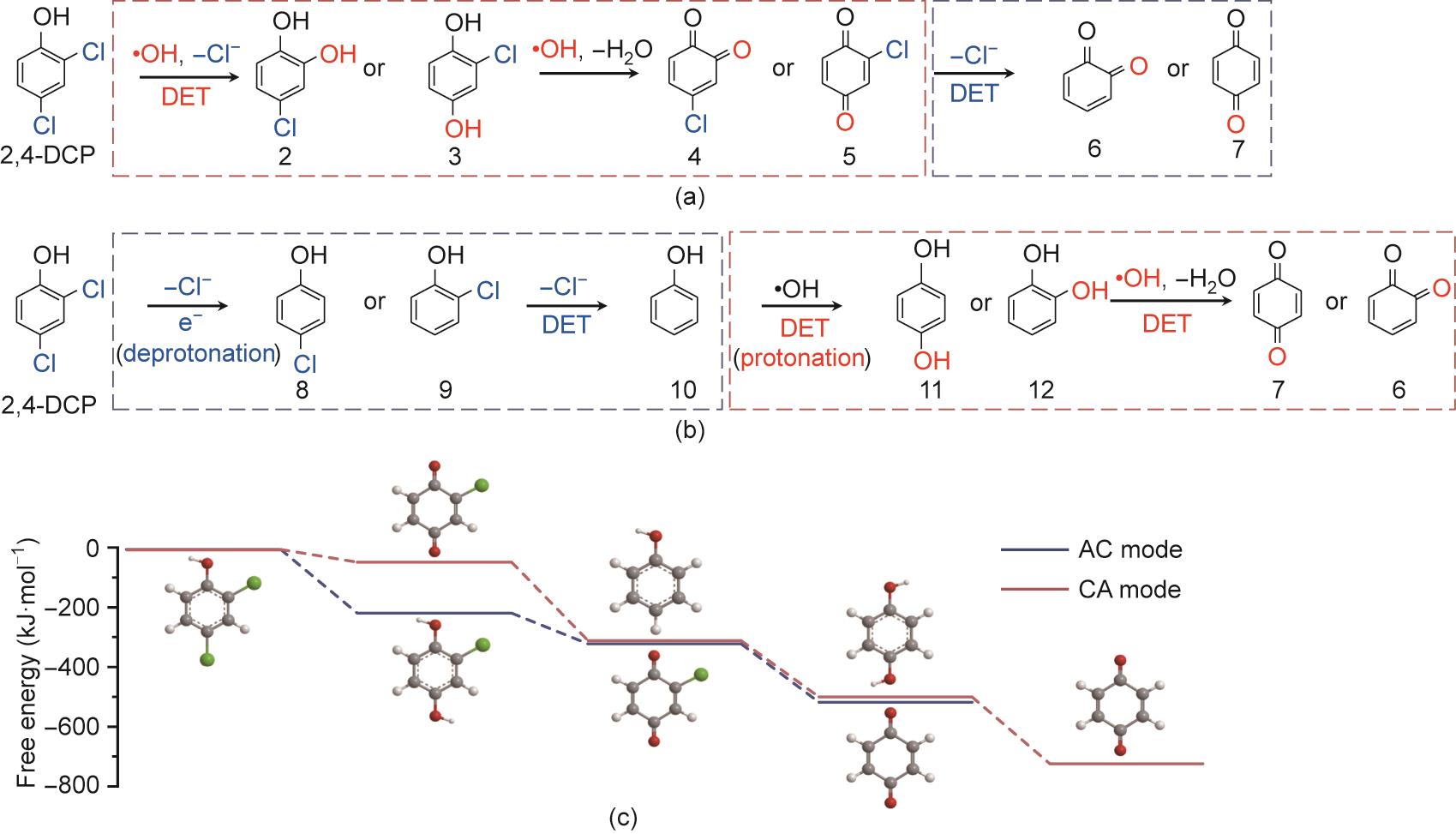
图4 AC(a)和CA(b)模式下的反应路径;(c)阳极和阴极电位分别为2.38 V vs SHE和-1.45 V vs SHE时2,4-DCP降解的DFT计算。AC:从阳极流向阴极;CA:从阴极流向阳极。
《3.5. REM组件放大策略》
3.5. REM组件放大策略
基于上述结果,本研究根据TCE构型放大穿流式电解反应器的规模。在电化学体系中,面积/体积比(A/V)即电极的比表面积(AS),是决定电化学体系固有降解能力的关键参数[36]。当REM反应器放大到较大规模时,AS显得格外重要。
如果电化学体系沿管式反应器(PPE)轴向放大,则在增加反应器尺寸和体积的同时必然导致AS降低,如下:
(11)
式中,L为电解池的轴向长度;r为平板电极的半径。虽然径向增加电极面积理论上也能放大PPE反应器,但这不仅增加了电极制备和工艺集成的复杂程度,还会导致电极上的一次电流分布不均匀[37]。
由于TCE构型允许水流穿过阳极侧壁,相较平板电极,圆柱状电极提供了更高的面积/体积比。通过增加TCE阵列数量可以简单地实现电解池的规模放大,其中,AS不再依赖于反应器的轴向长度和体积:
(12)
式中,r1为单个TCE组件的内管半径;R为反应器的半径;n为TCE组件数量。这种设计可以将任意轴向长度和数量的TCE阵列组件化,以便根据工程应用中需要处理的废水量来定制集成式电解反应器。
为了评估组件数量化策略的有效性,基于TiSO阳极设计了一个包含n = 1、2、3的TCE阵列电化学反应器[图5(a)]并考察了2,4-DCP降解的整体性能(10 mg·L-1)。由于面积/体积比随n值增加而增加,因此增加组件数量能够提升去除率和电流效率,并降低能量消耗,尤其在动力学较慢的AC模式下更为显著。在相同电流强度(4 A)下,将n值从1增加到3,使得AC模式下去除效率从27.3%增加到51.6%,CA模式下去除效率从92.5%增加到99.4% [图5(b)]。同时,AC模式下能量消耗(EEO)从24.6 kW·h·m-3降至10.8 kW·h·m-3,CA模式下从3.0 kW·h·m-3降至1.5 kW·h·m-3 [图5(c)]。此外,长期稳定的2,4-DCP去除效率证实了增加组件数量的放大策略具有可行性(附录A中的图S10)。尽管两种模式下电解池电压相似(n = 1、2、3时分别为4.7 V、3.8 V、3.5 V),但CA模式的能量消耗较AC模式高出近一个数量级。原因在于能量消耗的决定性因素是滤液出水浓度[Cp;方程(2)]而不是去除率,由于AC模式对有机污染物的破坏率较低,导致Cp较高,因而产生相当高的能耗。相应地,AC模式下的电流效率和COD去除率较CA模式分别低68.7%~87.0%和64.2%~87.9%(附录A中的图S11)。最低能耗1.5 kW·h·m-3时去除率仍可达99.4%,仍处于实验室规模的膜电极电化学高级氧化工艺能耗范围内(< 2.0 kW·h·m-3)[20]。效能更是高于传统电化学废水处理工艺,其中能量消耗高达每立方米几千瓦时(附录A的表S3 [37‒38]),主要是因为:产生·OH需要高达2.38 V vs SHE的热力学电位,以及电解水过程中伴随的析氧副反应。尽管如此,采用TCE的组件化工艺在处理分散式的大规模高盐度工业废水方面仍然具备竞争力。
《图5》

图5 (a)组件化TCE电解反应器的实物照片(n = 1, 2, 3);AC和CA模式下2,4-DCP的去除效率(b)和单位能耗(c)与组件数量的关系。AC:从阳极流向阴极;CA:从阴极流向阳极;进水溶液:[2,4-DCP] = 10 mg·L-1,总电流:4 A;流速为60 mL·min-1。
《4. 结论》
4. 结论
综上,本研究可以得出以下结论:亚氧化钛陶瓷阳极和不锈钢阴极组成的TCE构型适用于放大REM反应器规模。在TCE构型中,电极表面处处与电场线正交,欧姆电阻与电极长度成反比。TCE构型可以通过调节水流方向以AC或CA模式运行,从而为污染物的选择性降解创造条件。单程穿流式电解条件下,2,4-DCP的去除动力学常数在CA模式下较AC模式下高一个数量级,2,4-DCP和化学需氧量(COD)的去除率可分别达到98%和72.5%。TCE构型能够通过增加组件数量的策略放大电化学反应器规模,同时不会增加欧姆电阻或减少比电极面积。当采用三个TCE组件时,2,4-DCP的去除率可达99.4%,能耗为1.5 kW·h·m-3。本研究为REM反应器的规模化放大提供了一种合理、易于操作和可靠的策略,也能够推广应用于其他难降解有机污染物的去除和分散式电化学废水处理。











 京公网安备 11010502051620号
京公网安备 11010502051620号




