
《1、 引言》
1、 引言
含能材料是一类能够在一定外界刺激下,通过剧烈氧化还原反应释放出巨大能量的特殊反应性物质。自2000多年前中国发明黑火药以来,含能材料为人类的进步和繁荣做出了重大贡献[1‒2]。先进含能材料的能量、感度和热稳定性是最受关注的三个性能[3‒6]。然而,能量、感度和热稳定性之间始终存在着相互矛盾和制约的关系。一般来说,含能材料的高能量总是伴随着机械感度升高和热稳定性降低。因此,发展兼具高能量、低感度和良好热稳定性的新型含能材料仍然是一个巨大挑战。
为了指导含能材料的理论设计,人们已经发展出多种经验公式,如用于预测爆轰特性的Kamlet-Jacobs公式和用于预测机械感度的硝基电荷方法等[7‒8]。然而,这些经验公式很少能用于实验合成前的含能材料的大规模预筛选,原因是该类公式通常需要进行较为耗时的量子化学计算,而且其泛化能力也难以被确定。长期以来,新型含能材料的发现在很大程度上依赖于科学直觉及反复试错的过程[9],这种研发模式存在效率低、不确定性高等问题[10]。
随着大数据时代的到来,含能材料的研究范式发生了深刻变化[11‒12]。与经验模型相比,机器学习模型通常在准确性、泛化性和处理非线性问题的能力方面表现出优势[13],因此被广泛应用于材料科学的各个领域[14‒22]。在此,本文展示了一种机器学习辅助的高通量虚拟筛选(HTVS)系统,用于加速发现具有良好能量与安全性平衡的新型含能材料。该HTVS系统将机器学习模型与高通量分子生成相结合,从25 112个生成分子中快速筛选出性能优良的目标分子。筛选出的化合物能够表现出类石墨层状晶体堆积结构,这种特定的晶体堆积模式通常表现出更好的能量与安全平衡特性。经过对合成可行性的进一步评估,通过三步反应合成得到了一种性能较好的[5,6]稠杂环骨架基含能材料——7,8-二硝基吡唑并[1,5-a][1,3,5]三嗪-2,4-二胺(本文称为 ICM-104)。性能研究表明,含能材料ICM-104具有良好的综合性能,包括高能量、低感度和良好的热稳定性等。上述研究初步证明了所提出的HTVS系统的有效性以及机器学习在设计高性能含能材料方面的巨大潜力。
《2、 方法》
2、 方法
《2.1 数据准备与增强》
2.1 数据准备与增强
从过去几十年的文献中收集了1000多条含能材料数据,用于训练属性回归模型。该数据集包含具有多种结构的分子,涵盖脂肪族、芳香族、单环和多环化合物(有关详细样本和数据源请参见附录A中的数据集1)。附录A中的图S1提供了有关数据集的更多特征,如数据分布。在进行模型训练时,将所有数据以80∶20的比例随机分为训练数据和测试数据。将训练数据进一步分为训练集和验证集,用于进行五折交叉验证和调整超参数。五折交叉验证是指将验证集划分为5组,每组可用于一次验证,而其余4组用作训练集。最终测试分数是根据在训练过程中未使用的测试数据集计算而得。
为了训练分类模型,本研究从剑桥晶体学数据中心(CCDC)获取了365个被标记为“0”(表示不具有类石墨层状晶体堆积结构)的样本和22个被标记为“1”(表示具有类石墨层状晶体堆积结构)的样本(见附录A中的数据集2)。显然,现有数据量太小,不适合应用深度学习方法。因此,使用简化分子线性输入规范(SMILES)的枚举技巧进行数据增强,该技巧可以生成多个代表相同分子的不同SMILES字符串。SMILES枚举最早由Arús-Pous等[23]和提出,是一种用于分子深度学习的新型数据增强技术。标记为“0”和“1”的SMILES样本被分别放大了10倍和30倍。数据增强后,总样本量扩大到4000多个。在训练卷积神经网络(CNN)和长短期记忆(LSTM)模型时,保留400个样本作为测试集来评估模型的性能。
《2.2 特征与模型》
2.2 特征与模型
使用RDKit库提取了包括自定义描述符和电拓扑指纹在内的特征(即分子描述符)。属性预测模型通过Scikit-learn包中的核岭回归(KRR)算法进行训练。在KRR算法中,预测值(y*)可以表示为,给定一个核函数(k)[公式(1)]条件下,新样本(x*)与训练样本(x)内积的加权平均(α)。因此,学习过程中需要使用公式(2)计算系数矩阵(α,α为α的第i个元素),式中X、Y、λ和I分别为样本矩阵、标签矩阵、正则化参数和单位矩阵。使用网格搜索方法和五折交叉验证调整包括核函数在内的超参数。以决定系数R2 [公式(3),
(1)
(2)
(3)
(4)
分类模型中使用的CNN和LSTM是从Pytorch库中获取的。为了准备输入,从完整数据集包含的全部SMILES提取字典。字典的详细内容如下:[´N´, ´c´, ´1´, ´n´, ´(´,´)´, ´[´, ´+´, ´]´, ´=´, ´O´, ´-´, ´o´, ´2´, ´#´, ´C´, ´3´, ´H´, ´/´, ´\\´, ´4´, ´5´, ´None´](None用于填充)。因此,SMILES字符串被转换为大小为[120, 23]的二维(2D)数组。对于LSTM模型,SMILES的长度限制为120,允许出现的字符与字典的字符相同。此外,CNN包含两个2D卷积层和三个全连接层。2D卷积层的滤波器大小为16和32,而核尺寸均为7。最大池化层的核尺寸为2。全连接层的宽度分别为800、100和2。将整流线性单元(ReLU)作为激活函数。LSTM的隐藏层尺寸为64,层数为20。对于上述深度学习模型,损失函数均由交叉熵定义,并使用学习率为0.001的Adam优化器来更新权重。选择准确度[由公式(5)定义]、平衡准确度[由公式(6)定义]和F1分数[由公式(7)定义]作为评估模型性能的指标,其中TP、FP、TN、FN分别代表真阳性、假阳性、真阴性和假阴性。为了阐明采用深度学习算法的必要性,以基于描述符的K最近邻(KNN)作为基准进行测试。然而,SMILES枚举技巧并未被用于训练KNN模型,原因是由代表同一分子的不同SMILES所提取的描述符几乎完全相同。
(5)
(6)
(7)
(8)
(9)
为了对形成类石墨层状堆积结构的可能性进行评估,在预测过程也应用SMILES枚举技巧。对于代表同一分子的20个SMILES,经预测后可以得到类石墨层状堆积结构的比例(p)[公式(10)]。上述过程重复10次,以缓解由SMILES枚举的随机性造成的影响,并将p之和作为最终得分[式(11)]。
(10)
(11)
《2.3 制备及表征》
2.3 制备及表征
尽管本文涉及的化合物对外部机械刺激(如撞击和摩擦)的感度较低,但合成过程中使用了强腐蚀性浓硫酸。因此,建议在实验过程中使用防护手套、外套、面罩和防爆挡板等安全设备。
《2.3.1. 4-硝基-1-吡唑-3,5-二胺盐酸盐的制备》
2.3.1. 4-硝基-1-吡唑-3,5-二胺盐酸盐的制备
根据先前报道的路线[25]制备4-硝基-1H-吡唑-3,5-二胺。将浓盐酸(3 mL)加入4-硝基-1H-吡唑-3,5-二胺(3 mmol, 0.429 g)的甲醇(5 mL)悬浮液中。搅拌10 min后,过滤得到淡黄色固体,然后用乙酸乙酯(EtOAc)对其进行洗涤,得到4-硝基-1H-吡唑-3,5-二胺盐酸盐(产率为80%)。
《2.3.2. 8-硝基吡唑并[1,5-][,,]三嗪-2,4,7-三胺的制备》
2.3.2. 8-硝基吡唑并[1,5-][,,]三嗪-2,4,7-三胺的制备
该中间体是根据先前报道的路线略作修改[26]后制备的。首先,将4-硝基-1H-吡唑-3,5-二胺盐酸盐(3 mmol, 0.54 g)悬浮在无水乙醇(11 mL)中。然后,在悬浮液中加入双氰胺(4 mmol, 0.33 g)。将上述混合体系在80 ℃下回流6 h。在回流过程中,溶液中逐渐出现橙色固体。将橙色固体过滤并在80 ℃下用水重结晶,得到黄色固体(8-硝基吡唑并[1,5-a][1,3,5]三嗪-2,4,7-三胺;产率为60%)。
《2.3.3. 7,8-二硝基吡唑并[1,5-][,,]三嗪-2,4-二胺(ICM-104)的制备》
2.3.3. 7,8-二硝基吡唑并[1,5-][,,]三嗪-2,4-二胺(ICM-104)的制备
在冰水浴中,将8-硝基吡唑并[1,5-a][1,3,5]三嗪-2,4,7-三胺(3 mmol, 0.63 g)分批加入浓硫酸(6 mL)中,然后向溶液中滴加30%过氧化氢水溶液(2.5 mL)。在室温搅拌3 h后,使用碎冰淬灭反应,并使用乙酸乙酯萃取溶液。随后使用旋转蒸发仪除去乙酸乙酯,收集淡黄色固体即为目标化合物[7,8-二硝基吡唑并[1,5-a][1,3,5]三嗪-2,4-二胺(ICM-104);产率为42%]。目标化合物的核磁共振(NMR)数据如下所示。1H NMR (DMSO-d6, 400 MHz) δ: 8.81 ppm (s, 1H, NH2), 8.56 ppm (s, 1H, NH2), 8.04 ppm (s, 1H, NH2), 7.77 ppm (s, 1H, NH2);13C NMR (DMSO-d6, 100 MHz) δ: 162.41 ppm, 153.61 ppm, 150.44 ppm, 147.42 ppm, 109.47 ppm(见附录A中的图S12)。高分辨率电喷雾电离质谱(ESI-HRMS)数据如下所示。ESI-HRMS: m/z [M‒H]- 计算值为239.0283,测试值为239.0282(1)。红外光谱数据(IR; KBr, cm-1):3483.42, 3431.90, 3333.44, 3205.61, 1684.94, 1633.17, 1605.24, 1565.96, 1523.60, 1491.91, 1453.41, 1396.89, 1340.13, 1291.72, 1242.11, 1220.57, 1091.12, 983.45, 881.85, 851.93, 807.86, 784.96, 775.28, 728.80, 714.26, 600.36, 550.32。计算元素分析数值为:C 25.01%、H 1.68%和N 46.66%;实验元素分析结果为:C 24.67%、H 1.82%和N 46.40%。
1H和13C NMR数据通过Bruker(USA)Avance Neo 400 NMR核磁共振光谱仪收集,频率分别为400 MHz和100 MHz。使用具有电喷雾电离(ESI)的Shimadzu LCMS-IT-TOFTM质谱仪收集高分辨率质谱(HRMS)。使用标准BAM落锤和BAM摩擦测试仪进行撞击和摩擦感度测量。化合物的生成焓由燃烧热计算得到,燃烧热通过氧弹热量仪测量。使用Explo5(6.02版)软件计算标准爆轰性能。
《3、 结果与讨论》
3、 结果与讨论
《3.1 HTVS系统》
3.1 HTVS系统
HTVS系统的框架和组件如图1所示,具体功能及运行流程如图1(a)所示。首先,高通量分子生成模块可以根据输入母环及取代基迭代生成大量含能分子[图1(b)]。然后,将生成的分子导入属性预测器,进行快速准确的属性计算。属性预测器包含4个回归模型,以相同的复合分子描述符集作为输入,对密度、爆度、爆压和分解温度进行预测[图1(c)]。借助该属性预测器,可以根据预测的属性筛选具有较高能量、较低感度和良好热稳定性的潜在含能分子。然后将初步筛选出的、具有理想性能的分子送入晶体结构分类器,以进一步评估形成类石墨层状晶体结构的可能性。评估合成的可行性后,选择具有良好性能和较高概率形成类石墨层状晶体结构的分子进行实验合成和表征。该HTVS系统可以帮助研究人员通过分子生成和筛选过程定制含能材料,避免花费大量时间和精力进行实验试错。
《图1》
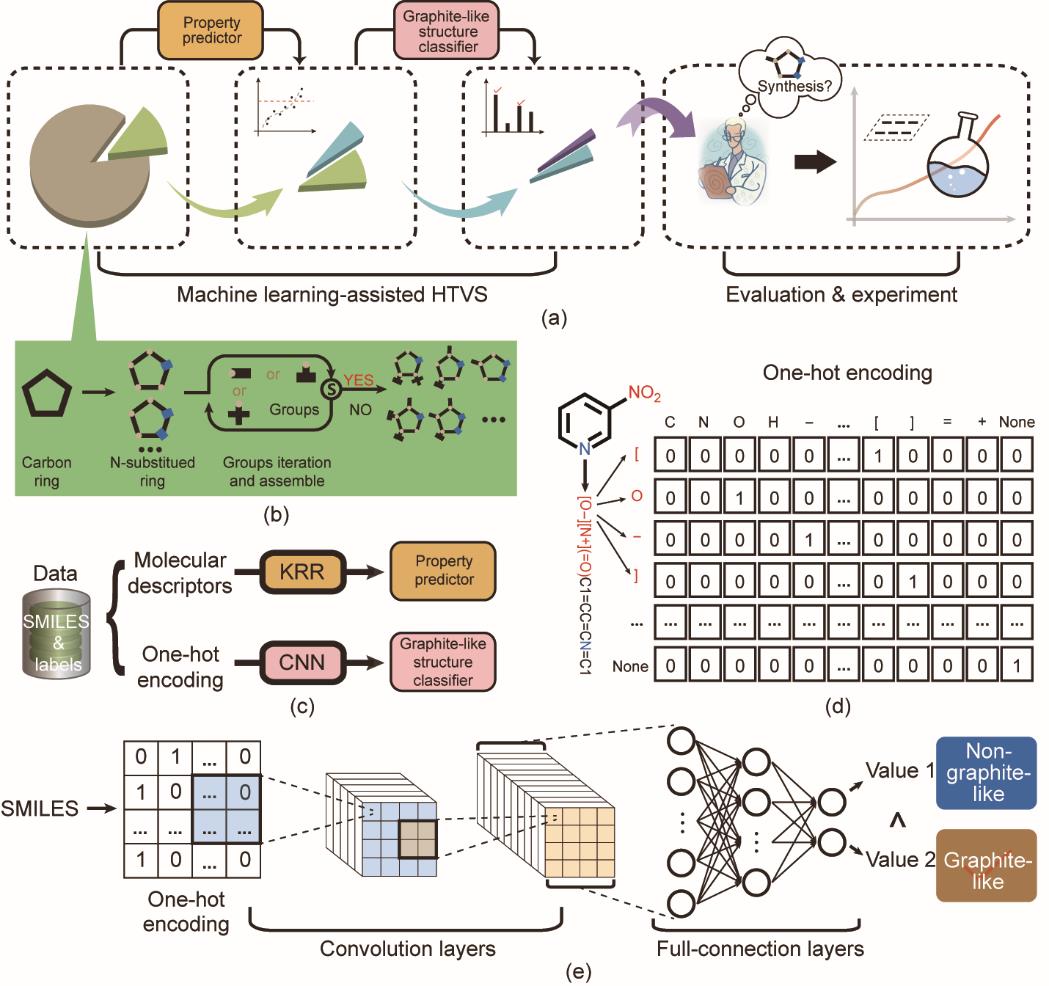
图1 HTVS系统的框架和组件。(a)机器学习辅助HTVS框架;(b)使用启发式枚举的分子生成示意图;(c)属性模型和类石墨层状堆积结构分类模型训练示意图;(d)CNN的one-hot输入编码;(e)CNN结构。
《3.2 特征集和属性模型》
3.2 特征集和属性模型
除了数据,特征(即分子描述符)是决定机器学习模型准确性的另一个重要因素。本研究采用的复合特征集(CDS)由两部分组成。第一部分为从电拓扑态(E-state)指纹谱中抽取的与碳(C)、氢(H)、氧(O)、氮(N)及卤素相关的指纹,该指纹谱已被广泛用于构建不同的模型来预测分子特性[27‒29]。另外,领域知识可以降低学习复杂性并提高特定任务的准确性。因此,本研究定义了一个自定义描述符集,其中包含另外的29个分子描述符(见附录A中的表S2)。此自定义描述符集增强了对分子形状和组成[如最佳拟合平面(PBF)和氧平衡(OB)]的描述,这将有助于对含能材料性质的学习。使用热力图可视化自定义描述符与密度数据的相关性[图2(a)],结果表明大多数自定义描述符没有显著相关性,这对于训练模型是有利的。
《图2》
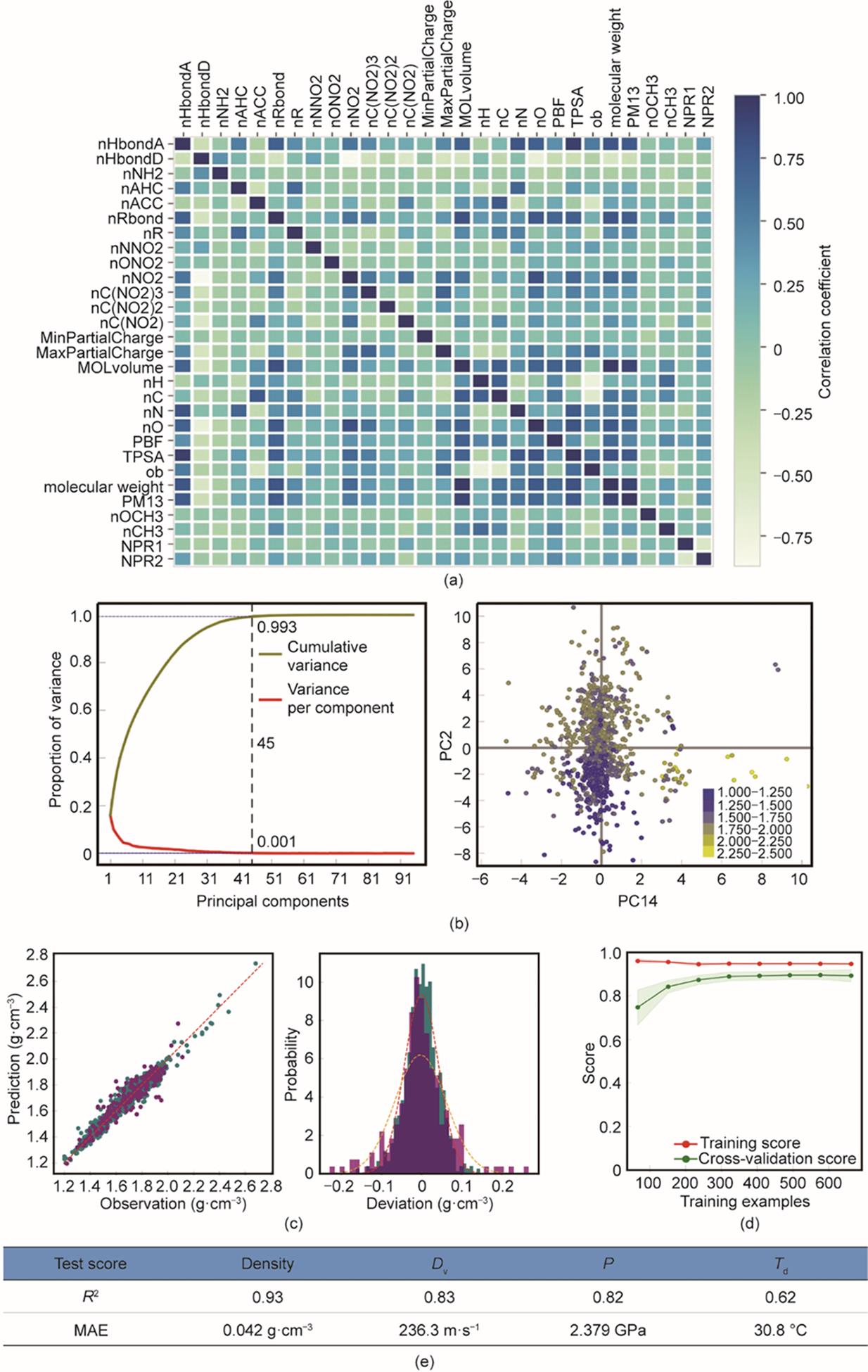
图2 性能预测模型的特征分布和模型评估。(a)自定义描述符集及其在密度数据上的特征分布热力图;(b)特征的PCA分析及密度数据上主要信息成分的散点图;(c)密度数据训练集(绿色)和测试集(紫色)的散点图和误差分布,其中红色(橙色)虚线是训练(测试)数据偏差的正态分布曲线;(d)密度训练模型的学习曲线(红色为训练曲线,绿色为交叉验证曲线);(e)4个训练模型的测试分数(Dv:爆速;P:爆压;Td:分解温度)。
通过主成分分析(PCA)法分析CDS在密度数据中捕获基础模型的能力[30]。当将原始特征组合成45个主成分时,累积方差达到0.993 [图2(b),左]。此外,通过对主要成分(PC14和PC2)信息最丰富投影进行可视化[图2(b)],可以看到不同密度的样本分布相对集中,并观察到明显的颜色梯度,这意味着这些特征能够有效地刻画密度数据的潜在模型。
在使用KRR算法[31]训练模型后,分别通过比较训练集和测试集上的观察值和预测值来验证模型预测密度的性能[图2(c)]。结果发现,观察值和预测值之间存在显著的一致性[图2(c)],并且它们之间的偏差符合正态分布[图2(c),右]。在学习曲线中,随着训练样本的增加,训练曲线(红色)和交叉验证曲线(绿色)都逐渐接近相同的渐近线[图2(d)],说明本文的模型被训练得很好(即没有观察到过拟合或欠拟合)。测试数据集的决定系数(R2)和MAE分别为0.93 g∙cm-3和0.042 g∙cm-3 [图2(e)]。密度模型的高精度可能源于大量的数据和合理的特征化方法,可以在一定程度上捕捉分子和晶体的特征。以相同的复合分子描述符集作为输入,对爆速(Dv)、爆压(P)和分解温度(Td)的预测模型进行训练。如图2(e)所示,Dv、P和Td模型在测试数据集上测试的R2值分别为0.83(MAE: 236.3 m∙s-1)、0.82(MAE: 2.379 GPa)和0.62(MAE: 30.8 ℃)。对于这些模型的训练和评估,请参见附录A中的图S2;交叉验证分数和训练稳定性测试的更多结果见附录A的表S3。值得注意的是,与过去的工作相比,本文的模型在准确性、有效性和全面性方面更具有竞争力(见附录A中的表S4)。除了上述4个性能(密度、爆速、爆压和分解温度)外,感度也是含能材料的核心性质。但目前训练通用的感度预测模型仍然很困难,主要原因是感度与包括电子结构、晶体结构甚至测量条件在内的多尺度因素相关。因此,亟需一种解决感度预测问题的替代方法。
《3.3 类石墨层状晶体结构的分类模型》
3.3 类石墨层状晶体结构的分类模型
为了找到一种更可靠的方法来快速筛选具有低感度的含能分子,本研究尝试将撞击感度的直接预测转化为类石墨层状晶体堆积模式的识别,原因是一般类石墨层状晶体结构和含能材料的低感度之间存在显著相关性[32‒34]。晶体结构与分子结构存在联系,特别是某些倾向于形成强非键相互作用的官能团可能主导晶体的形成。在之前的一些研究中,深度神经网络被用于预测晶体结构,这启发了本研究采用深度学习来帮助解决这一问题[35‒36]。
基于上述考虑,本文选择CNN和LSTM [37‒38]来捕捉由分子结构推断能否形成类石墨层状晶体结构的化学直觉。CNN使用分子SMILES字符串的one-hot编码作为输入进行训练[图1(c)、(d)] [39‒40],其网络结构如图1(e)所示。LSTM直接使用SMILES作为输入进行训练。此外,对使用CDS作为输入的KNN模型(CDS + KNN模型)进行训练,并将其作为基准,与深度学习模型进行比较。训练过程的比较如图3所示,结果表明SMILES_Onehot + CNN模型优于SMILES + LSTM模型,原因是前者的训练和测试损失较低,并且前一个模型的精度及平衡精度高于后一个模型。通过混淆矩阵可以发现具有最低测试损失的SMILES_Onehot + CNN(epoch 15)模型的表现比SMILES + LSTM更好,因为后者更倾向于将类石墨层状堆积分子(“1”)误分类为非石墨层状堆积(“0”)分子。相比之下,从平衡精度(0.65)和混淆矩阵方面看,CDS + KNN模型表现出较差的精度。出现这种结果的主要原因是CNN和LSTM模型中保留了更多关于分子结构的信息(如原子和取代基团的排列;这对于预测晶体堆积至关重要),而在CDS + KNN模型中,这些信息在特征化过程中被压缩损失掉了。本研究还尝试了更简单的架构(如基于CDS的决策树和神经网络,结果见附录A中的表S5),结果表明SMILES_Onehot + CNN模型在准确性方面表现出绝对优势。
《图3》

图3 分类模型的比较。(a)SMILES_Onehot + CNN模型的训练过程和混淆矩阵;(b)SMILES + LSTM模型的训练过程和混淆矩阵;(c)CDS + KNN模型的混淆矩阵;(d)测试数据上的模型评价指标。
最后,将SMILES_Onehot + CNN模型与SMILES枚举技巧相结合,以评估潜在分子具有类石墨层状晶体结构的可能性[41]。可能性值表示一个分子形成类石墨层状堆积结构的趋势;该方式有利于按照从高到低的可能性对这些分子进行分类和评估。通过上述方式,类石墨状层状晶体结构的筛选步骤变得更加稳健。
《3.4 含能分子的高通量生成和筛选》
3.4 含能分子的高通量生成和筛选
按照启发式枚举的思路通过自制脚本(见附录A中的图S3)进行分子生成[图1(b)] [42‒43]。近年来,研究人员对氮杂稠环含能分子(如[5]稠杂双环和[5,6]稠杂双环含能)表现出越来越大的兴趣,相关研究已经报道了一系列有前景的稠环含能分子[44‒48]。本研究重点关注了由[5,6]稠杂双环骨架和硝基/氨基构成的含能分子。
分子生成的初始输入结构包含5个不同的[5,6]双碳环。经过氮取代(从1个氮到7个氮)过程之后,获得了355个不同的[5,6]稠杂双环骨架[图4(a)]。考虑到分子生成耗时和实验合成的可行性,[5,6]稠杂双环骨架中的最多取代基位点被限制为4个(见附录A中的图S2中的散点图)。将硝基/氨基引入355个不同的[5,6]稠杂双环骨架中,在结构整理和去重后共计生成了25 112个[5,6]稠杂双环分子。如附录A中的图S4所示,生成分子的性能分布与训练数据涵盖范围相符。
《图4》
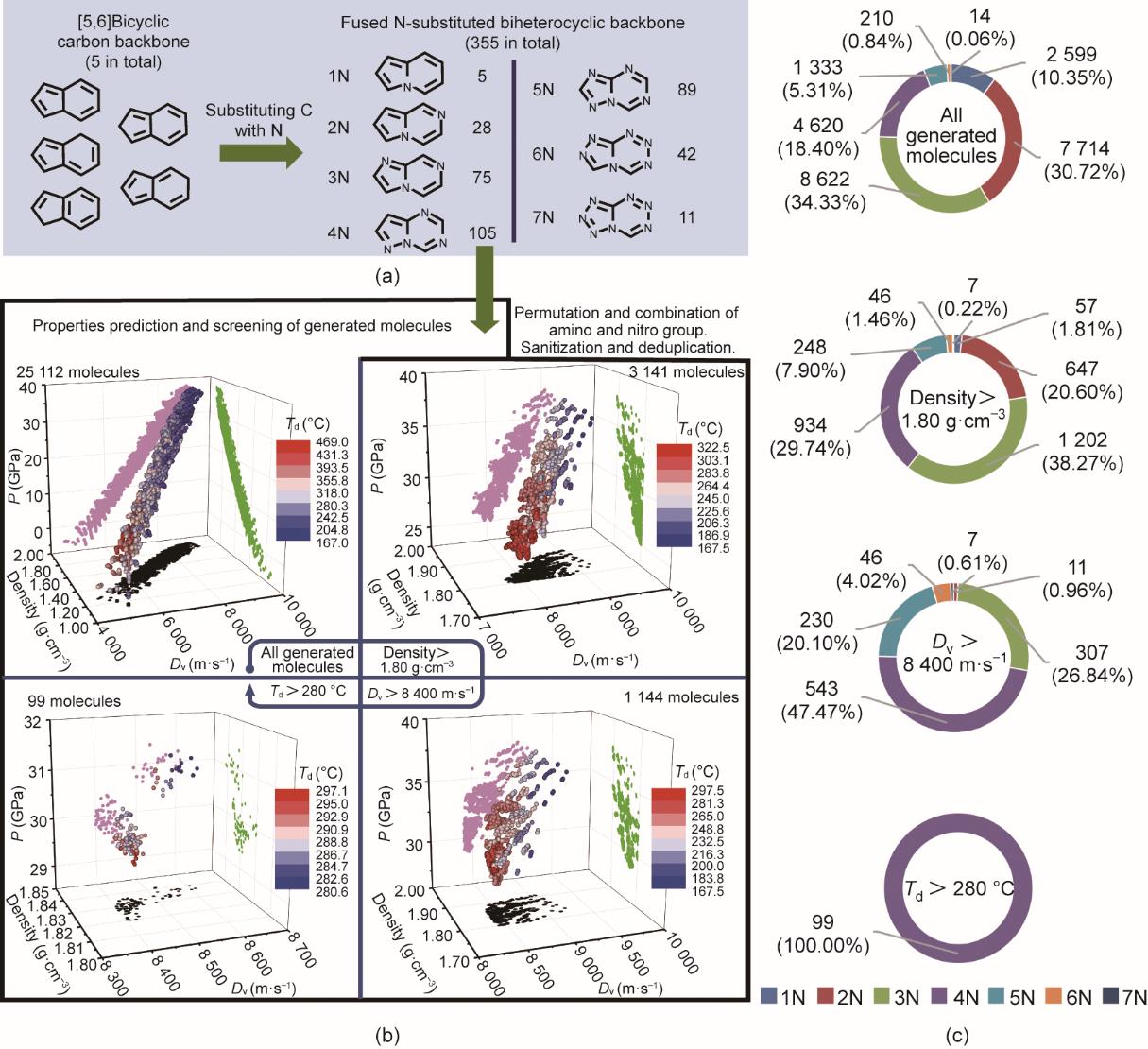
图4 分子生成及筛选过程。(a)[
随后将生成的25 112个含能分子输入属性预测器中,以预测它们的属性(包括密度、Dv、P和Td),并进行筛选(见附录A中的补充数据1)。借助三维(3D)填色散点图[图4(b)]和环状图[图4(c)]对整个分子空间和逐步筛选过程进行可视化。25 112个分子的预测属性符合含能材料的一些一般规律,如密度和Dv/P之间的线性相关性。密度与和分解温度之间呈负相关[图4(b)]。将经典含能材料环三次甲基三硝基胺(1,3,5-trinitro-1,3,5-triazinane, RDX)的密度(1.80 g∙cm-3)作为筛选的第一个标准,筛选后,分子数量从原来的25 112个急剧减少到3141个[图4(b)]。3D填色散点图表明,Td高于280 ℃的分子(红点)大多位于Dv值相对较低的区域(约8000 m∙s-1)。而Dv大于8800 m∙s-1的分子(蓝点)大多位于Td值相对较低的区域(约160 ℃)[图4(b)]。当分别引入能量(Dv > 8400 m∙s-1)和热稳定性(Td > 280 ℃)筛选标准(见附录A中的图S5)时,满足要求的分子数量从3141个减少到1144个 [图4(b)]。最后,只有99个分子满足全部筛选条件[见图4(b)和附录A中的图S6]。
随着筛选标准的逐步引入,环状图清楚地显示了不同氮取代的[5,6]稠杂双环分子比例的变化[图4(c)]。引入密度(> 1.80 g∙m-3)和能量(Dv > 8400 m∙s-1)的筛选标准后,5个(天蓝色)、6个(橙色)和7个(深蓝色)氮原子取代的[5,6]稠杂环分子比例分别从5.31%、0.84%和0.06%增加到20.10%、4.02%和0.61%,这意味着分子骨架中的高氮含量有利于增加分子的能量(高密度和高Dv值)。但是,高氮含量会降低分子的热稳定性,导致分解温度难以超过280 ℃。相比之下,1个(蓝色)和2个(红色)氮原子取代的[5,6]稠杂环分子的比例分别从10.35%和30.72%下降到0和0.96%,低于密度及爆速的筛选标准,表明低氮含量对分子能量的提升是不利的。通过密度(> 1.80 g∙cm-3)和能量(Dv > 8400 m∙s-1)筛选后,在筛选出的1144个候选化合物中,3个氮取代的[5,6]稠杂双环分子(绿色)显示出相对较高的百分比(26.84%)。然而,它们的分解温度不能满足高热稳定性(Td > 280 ℃)的标准,主要是因为3个氮取代的[5,6]稠杂双环分子的氮含量仍然相对较低,导致满足密度和能量标准的筛选分子通常含有多个硝基(一般含有3个或4个)(见附录A中的图S7);但多个硝基的强吸电子作用会降低分子稳定性,从而导致其难以满足分解温度筛选标准(Td > 280 ℃)。总之,经过三步筛选,最后留下的99个分子均为4个氮原子(紫色)取代的[5,6]稠杂双环分子;从分子稳定性的角度看,含4个氮原子的稠环分子(4N)中的氮含量和硝基的数量都较为合理。
将这99个含能分子导入类石墨层状堆积结构分类器,对它们形成特殊类石墨层状晶体结构可能性进行打分。对每个分子的预测重复5次,结果见图5(a)和附录A中的数据集2。根据平均分数从高到低进行排序,前5个分子结构如图5(b)所示。在评估了这5个分子的合成可行性(见附录A中的图S8)后,发现分子2 [图5(b);7,8-二硝基吡唑并[1,5-a][1,3,5]三嗪-2,4-二胺,在此命名为ICM-104]从未被报道过,并且具有较高合成可行性。因此,选择分子2作为目标分子进行后续实验。
《图5》

图5 可能形成类石墨层状晶体结构的分数。(a)99个候选化合物形成类石墨层状晶体结构的平均分数(误差线表示5次预测的平均偏差);(b)可能性排名前五的分子结构。
《3.5 合成及性能研究》
3.5 合成及性能研究
令人鼓舞的是,根据设计的合成路线,通过三步反应成功制备了目标分子ICM-104(第2.3节)。将其饱和的乙酸乙酯溶液进行缓慢溶剂挥发,获得适合X射线衍射的ICM-104单晶(见附录A中的表S6)。ICM-104具有类石墨层状晶体堆积结构,与预期结构一致,空间群为P21/c[图6(a)]。在分子结构中,一个硝基在超分子平面之外(夹角为66.7°),这是由相邻的两个硝基相互排斥作用造成的[图6(a)]。ICM-104的超分子平面由氨基、硝基和氮原子之间的氢键构成[图6(a)]。这一结果表明,训练后的类石墨层状堆积结构分类模型有助于识别具有独特类石墨层状晶体堆积的新型含能分子。
《图6》

图6 ICM-104的晶体结构和性质。(a)ICM-104的3D类石墨层状晶体堆积、2D超分子平面和分子几何结构;(b)ICM-104、2,4,6-三氨基-1,3,5-三硝基苯(TATB)和2,6-氨基-3,5-二硝基吡嗪-1-氧化物(LLM-105)的预测值与实测/计算性能之间的比较[黑绿色代表通过实验测量或使用Explo5(v6.02)计算的特性,而淡紫色代表所提出的机器学习模型预测的性能];(c)ICM-104、LLM-105和TATB(1 kcal = 4.19 × 103 J)的硝基电荷、最大静电势(ESP)和电荷平衡的比较;(d)ICM-104、LLM-105和TATB层间相对滑动的能量变化,其中深黄色表示选择的滑动分子层。
完成ICM-104的结构表征之后,通过将实验/计算结果与使用模型预测的结果进行比较来评估预测模型的实用性。如图6(b)所示,ICM-104的预测密度、Dv和P分别为1.828 g∙cm-3、8422 m∙s-1和29.8 GPa [图6(b)中的绿色直方图],接近实验密度(1.825 g∙cm-3)和计算的Dv值和P值[8551 m∙s-1和29.8 GPa;使用Explo5(v6.02)获得] [图6(b)中的淡紫色柱状图]。分解温度(Td)的实验值(326 ℃)和预测结果(286 ℃)之间存在约40 ℃的偏差。造成这种偏差的主要原因是ICM-104的晶体是由强分子间氢键构筑而成,而本研究目前的复合描述符集主要集中在分子水平上,描述分子间相互作用的能力相对较弱。ICM-104的分解温度高达326 ℃(见附录A中的图S9),与2,6-氨基-3,5-二硝基吡嗪-1-氧化物(LLM-105)的分解温度(342 ℃)及2,4,6-三氨基-1,3,5-三硝基苯(TATB)的分解温度(350 ℃)接近。使用Kissinger和Ozawa方法获得的ICM-104的非等温动力学表观活化能(Ea)分别为615 kJ∙mol-1和594 kJ∙mol-1(见附录A中的图S9),表明ICM-104具有优异的热稳定性。ICM-104较高的分解温度归因于其类石墨层状堆积结构,这种堆积模式有利于实现更好的热稳定性和相对更高的引发键键能[键解离焓为260.63 kJ∙mol-1,较LLM-105(247.72 kJ∙mol-1)更高;见附录A中的图S10] [49]。此外,ICM-104还表现出较低的撞击(测量值为35 J)和摩擦(测量值高于360 N)感度。同时,TATB(1.882 g∙cm-3、7964 m∙s-1、26.8 GPa和317 ℃)和LLM-105(1.906 g∙cm-3、8537 m∙s-1、31.5 GPa和289 ℃)的预测值接近于它们的实测/计算结果[图6(b)]。通过详细的实验评估以及与TATB和LLM-105的性能比较[见图6(b)和附录A中的表S7],可以发现ICM-104是一种很有前景的耐热不敏感含能材料。
本文从分子结构和晶体堆积方式两个层次定性地阐述了ICM-104表现出较低机械感度的原因。包括硝基电荷、最大静电势(ESP)和电荷平衡(上述参数使用Gaussian 09 D.01和Multiwfn 3.7计算)在内的三个分子层面的参数常用于评估分子在机械刺激下的稳定性[50‒51]。如图6(c)所示,从分子层面看,在这三种化合物中,TATB无疑具有最低的感度。进一步将LLM-105与ICM-104进行比较,LLM-105的ESP最大值和电荷平衡(分别为44.6 kcal∙mol-1和0.243)优于ICM-104(分别为60.7 kcal∙mol-1和0.219)。虽然LLM-105(-0.393e)的硝基电荷略高于ICM-104(-0.485e)[52],但可以认为LLM-105的分子结构比ICM-104更稳定。另一方面,采用力场方法计算了层间相对滑动过程可能导致的能量变化,以评估晶体堆积对感度的贡献。如图6(d)所示,能量变化的强度按照LLM-105 > ICM-104 ≫ TATB的顺序降序排列。在对抗外部机械作用时,类石墨层状堆积结构的ICM-104比波浪状晶体结构的LLM-105具有更好的缓冲作用。然而,扭曲的硝基可能会在滑动过程中引发层间产生强烈的排斥力。因此,ICM-104对应的能量变化仍然比TATB更剧烈。基于上述分析,ICM-104表现出的机械感度介于LLM-105和TATB之间是合理的。如附录A中的图S10所示,通过与最近报道的稠环化合物的性能比较,可以进一步凸显ICM-104的综合性能优势。在最近的工作中,本文提出的机器学习辅助HTVS系统还被应用于探索含能熔铸材料[53]。总体而言,本研究所建立的机器学习辅助HTVS系统在指导发现具有所需结构和性能的新型含能材料方面表现出巨大潜力。
《4、 结论》
4、 结论
本研究开发了一个机器学习辅助的HTVS系统,并用于指导含能材料的探索。该HTVS系统集成了高通量分子生成和机器学习模型。高通量分子生成模块负责通过启发式枚举快速、全面地生成需求的分子结构。机器学习模型由属性预测器和类石墨层状堆积结构分类器组成。属性预测器包含4个回归模型(包括密度、爆速、爆压和分解温度),而结构分类器源自CNN分类模型,能够对形成类石墨状层状结构的可能性进行评估。基于HTVS系统,从25 112个[5,6]稠杂双环分子中迅速发现了具有优秀性能的ICM-104。进一步的实验研究表明,ICM-104表现出与预期相符的良好性能,包括良好的爆轰性能(密度为1.825 g∙cm-3、Dv = 8551 m∙s-1、P = 29.8 GPa)、低感度(撞击感度为35 J,摩擦感度为360 N)和良好的热稳定性(初始分解温度为326 ℃)。本研究证明了机器学习辅助HTVS系统在快速发现新型含能材料方面的潜力。此外,本文所提出的系统方法可以被用于发现其他有机功能材料。











 京公网安备 11010502051620号
京公网安备 11010502051620号




