

《1、 引言》
1、 引言
非伤寒沙门菌(non-typhoidal Salmonella enterica, NTS)是全球重要的食源性致病菌。世界卫生组织(WHO)在《全球食源性疾病负担报告》中估计,NTS导致的胃肠炎疾病负担约为400万伤残调整寿命年,是导致食源性疾病死亡的主要原因[1]。鼠伤寒沙门菌(S. Typhimurium)是导致NTS病的常见血清型。然而近年来,一种多重耐药(multidrug resistant, MDR)单相鼠伤寒沙门菌(S. 4,[5],12:i:-)在全球广泛流行[2],该血清型缺少鼠伤寒沙门菌fljB编码的二相鞭毛抗原(抗原式为4,[5],12:i:-)。自20世纪80年代末首次被发现以来,S. 4,[5],12:i:-逐渐成为全球NTS病中最常见的血清型之一[3‒4]。该血清型在猪肉生产链中广泛流行,与人类感染密切相关。WHO于2022年3月27日报道了一起由S. 4,[5],12:i:-污染巧克力产品引发的多国感染暴发事件,提示该致病菌可经其他食物载体传播[5]。
既往研究报道了S. 4,[5],12:i:-存在三个克隆群(西班牙克隆、美国克隆和欧洲克隆),具有不同的噬菌体型(phage type, PT)和耐药谱。“西班牙克隆”于1997年在西班牙发现,噬菌体型为PT U302,序列型(sequence typing, ST)为ST19,较鼠伤寒沙门菌LT2缺失5个区域[6],该克隆携带的质粒介导了对氨苄青霉素、氯霉素、庆大霉素、链霉素/奇霉素、磺胺类、四环素和甲氧苄啶7类抗生素耐药。“美国克隆”出现于2004年,与“西班牙克隆”的主要区别在于fljBA操纵子周围的基因发生缺失,但hin、iroB及STM1053-1997区域没有变化(“西班牙克隆”均缺失)[3]。此外,“美国克隆”中较少有多重耐药的菌株(主要是ST19,对氨苄西林、链霉素、磺胺类和四环素均敏感)。2005年发现的“欧洲克隆”的主要噬菌体型为PT DT193和PT DT120,序列分型为ST34,但鼠伤寒沙门菌的典型毒力质粒pSLT缺失。欧洲菌株具有染色体编码的blaTEM-1、strAB、sul2和tet(B)耐药基因,介导ASSuT耐药表型,这些基因可分布在两个不同的基因环境[耐药区域(resistant region, RR)1和RR2],已在欧盟部分国家迅速蔓延[7‒9]。此外,泛耐药ST313 S. 4,[5],12:i:-在非洲出现并导致持续暴发[10]。
自2000年以来,全球肉类产量急速增长50%,在中低收入国家尤为明显,未来可能将持续增长。对动物蛋白的需求也将导致动物养殖中抗生素的使用量持续增加[11],若动物生产中滥用抗生素则会导致多重耐药菌的出现。动物食品中已出现了多重耐药S. 4,[5],12:i:-,其快速传播能力对人类健康及全球公共卫生安全构成了严重威胁[12],亟待加以控制。
近十年来S. 4,[5],12:i:-在中国的流行率不断上升,其多重耐药性也引起广泛关注。中国动物源分离株多来自肉猪,对四环素、氨苄青霉素、磺胺异𫫇唑、链霉素、萘啶酸、阿莫西林-克拉维酸、氯霉素的耐药率较高[13‒14]。猪肉和患者来源的S. 4,[5],12:i:-分离株的主要序列型为ST34 [15‒17]。中国是全球最大的猪肉生产国和消费国,此外,全球贸易(食用动物、食品)和人员流动是感染性疾病传播的潜在因素。目前,欧洲、美国和澳大利亚流行的S. 4,[5],12:i:-基因组特征已基本明确[7‒9,12],但在中国流行的该血清型菌株的基因组特征及谱系进化关系报道较少,因此深入研究该血清型的分子流行病学对未来新发食源性疾病的防控至关重要。
本研究重点分析了2008—2017年河南省腹泻患者分离的100株S. 4,[5],12:i:-的耐药性和基因组流行病学特征。基于抗菌药物敏感性试验和全基因组测序(whole genome sequence, WGS),阐明了S. 4,[5],12:i:-在中国的流行特征及耐药机制;进一步与其他国家来源菌株进行比对分析,明确了中国特有谱系及耐药谱差异的遗传进化的潜在驱动力。综上,腹泻患者来源的菌株为研究不同谱系进化和阐明动物源致病菌在耐药性传播方面的作用提供良好的资源。
《2、 材料和方法》
2、 材料和方法
《2.1 菌株分离与鉴定》
2.1 菌株分离与鉴定
2008—2017年,从河南省6个地区腹泻患者分离的1565株沙门菌菌株中共鉴定获得100株S. 4,[5],12:i:-(附录中的表S1)[18]。患者样品收集方法详见已发表文献[19],采用API 20E(bioMérieux,中国)和invA基因对分离株进行沙门菌鉴定[20]。针对S. 4,[5],12:i:-菌株,通过玻片凝集法确定血清型,再依据欧盟食品安全局(EFSA)的多重聚合酶链反应(PCR)法进行确证[21]。
《2.2 抗菌药物敏感性试验》
2.2 抗菌药物敏感性试验
按美国临床标准委员会(CLSI)推荐的标准[22]和欧洲抗菌药物敏感性试验委员会(EUCAST)[23]规程,采用琼脂稀释法对100株S. 4,[5],12:i:-分离株开展15种抗菌药物[氨苄西林(AMP)、头孢噻肟(CTX)、头孢他啶(CAZ)、亚胺培南(IPM)、美罗培南(MEM)、庆大霉素(GEN)、阿米卡星(AMI)、链霉素(STR)、磺胺异𫫇唑(SUL)、复方新诺明(SXT)、氯霉素(CHL)、四环素(TET)、萘啶酸(NAL)、环丙沙星(CIP)和黏菌素(CT)]的敏感性试验。以大肠杆菌ATCCTM 25922和肺炎克雷伯菌ATCCTM 700603为质控菌株。MDR指对三种或以上不同类别的抗菌药物耐药。
《2.3 全基因组测序、从头组装和注释》
2.3 全基因组测序、从头组装和注释
采用Wizard®试剂盒(Promega, Madison WI,美国)提取细菌基因组DNA,经凝胶电泳检测质量及Qubit2.0(Invitrogen, Life Technologies,美国)定量。细菌基因组DNA经Illumina NovaSeq 6000平台(Illumina,美国)测序。所构建DNA文库片段平均长度为500 bp,基于150 bp的双端测序策略进行测序,测序深度为80×。过滤去除低质量读段,利用SPAdes v3.13 软件进行基因组组装[24]。对6株代表性分离株基于MinION测序平台进行第三代纳米孔测序(ONT,英国),使用Unicycler软件进行混合组装[25]。通过Sanger测序法和MinION对复杂质粒结构进行确认[26]。使用prokka软件进行全基因组基因预测和注释[27],基于BLASTn软件同ResFinder、ISFinder、VFDB和PlasmidFinder数据库进行比对,分析菌株的耐药基因、插入序列、毒力基因和复制子类型[28‒31]。
《2.4 基于核心基因组单核苷酸多态性的系统发育分析》
2.4 基于核心基因组单核苷酸多态性的系统发育分析
从美国国家生物技术信息中心(NCBI)数据库下载标准菌株鼠伤寒沙门菌LT2(GenBank序列登录号:NC_003197)、2017年河南省养猪场猪粪便样品中分离的4株S. 4,[5],12:i:-(GenBank项目登录号:PRJNA573539)及截至2019年收录的345株欧洲来源和209株美国来源的ST34型S. 4,[5],12:i:-的基因组序列,以LT2为参考序列,使用bowtie2软件进行原始测序读段数据比对,使用Samtools软件进一步分析获得单核苷酸多态性位点(single nucleotide polymorphisms, SNPs)。过滤距离5 bp内的相邻突变以去除由重组造成的变异位点,最终获得5条及以上读段支持的高质量核心基因组SNP位点[32‒34]。将各菌株中核心基因组SNP位点的碱基串联,使用FastTree v2.1.10软件基于最大似然法进行系统发育分析[35],利用iTOL软件进行可视化,获得系统发育关系和遗传性状(耐药基因等)携带情况[36]。
《2.5 基于贝叶斯法的分化时间估计》
2.5 基于贝叶斯法的分化时间估计
采用BEAST v1.8.2软件,基于贝叶斯模型进行系统发育分析[37]。基于菌株的分离年份在构建系统发育关系的同时估计种群分化时间、描述种群动态变化。选用不同分子钟模型和树先验模型进行比较分析,数据集的最佳拟合进化模型选择Hasegawa-Kishino-Yano (HKY)核苷酸替换模型,位点间异质性的先验分布使用正态分布模型。选用严格分子钟或宽松分子钟模型以及Coalescent Constant Population或Coalescent Exponential Population先验模型。
《2.6 核苷酸序列登录号》
2.6 核苷酸序列登录号
菌株2008079、2010112、2016062、2016089、2017005和2017028的全基因组序列已提交NCBI,序列登录号分别为CP090529~CP090546。全基因组测序原始数据已提交NCBI(GenBank项目登录号:PRJNA794010)。
《3、 结果》
3、 结果
《3.1 菌株信息及耐药谱》
3.1 菌株信息及耐药谱
2008—2017年河南省6个地区1565株腹泻患者来源的沙门菌信息见表S1。其中,经血清分型和多重PCR鉴定出100株S. 4,[5],12:i:-(6.4%, 100/1565),为第三位最常见血清型(表S1)。在本项目的研究期间,S. 4,[5],12:i:-分离率快速上升,各年度流行率为0.6%~15.6%(图1)。
《图1》

图1 (a)104株S. 4,[
100株S. 4,[5],12:i:- 除对亚胺培南和美罗培南敏感外,对其他13种抗生素均有不同程度的耐药,97株是MDR (对三种或以上不同类别的抗菌药物耐药,见附录A中的表S2)。对不同抗生素的耐药率从高到低依次为四环素(98.0%)、磺胺类(97.0%)、氨苄青霉素(91%)、链霉素(77.0%)、氯霉素(61.0%)、萘啶酸(56.0%)、环丙沙星(53.0%)、复方新诺明(52.0%)(附录A中的表S3);对头孢噻肟的耐药率(13.0%)较低,但有12株分离株同时对环丙沙星也耐药;对黏菌素(3.0%)和阿米卡星(1.0%)耐药的分离株较少见。多数分离株(73.0%)具有ASSuT耐药谱,其中,42株还对氯霉素耐药。与欧洲和美国所报道的分离株显著不同的是,中国分离株对氟喹诺酮类、氨基糖苷类和氯霉素类耐药性较高(附录A中的表S3、表S4)[7‒8,12]。
《3.2 耐药基因分布及突变情况》
3.2 耐药基因分布及突变情况
基于WGS数据,100株分离菌均鉴定为ST34 S. 4,[5],12:i:- [38]。从WGS数据中筛选已知抗生素耐药基因(antibiotic resistance genes, ARGs),包括水平转移基因和染色体上喹诺酮耐药决定区(quinolone resistance-determining region, QRDR)基因突变。共检出35种耐药基因,并存在gyrA基因点突变。中国分离株耐药基因携带情况与欧美菌株有明显差异[图1(a)、图2、图3]:半数以上的分离株携带6类抗生素的耐药基因,包括blaTEM-1B(n = 65,氨苄青霉素耐药)、strA和strB(n = 71,链霉素)、sul1(n = 53,磺胺类药物)和sul2(n = 79,磺胺类药物)、tet(B)(n = 98,四环素)、floR(n = 51,氯霉素)及arr-3(n = 50,利福平);此外,49株分离株携带dfr等位基因(甲氧苄啶),包括dfrA12 (n = 38)和dfrA27 (n = 11)两个变异体;39株分离株携带编码的氯霉素耐药基因catB3;39株分离株检测到三种gyrA点突变导致的氨基酸替换,包括D87N (n = 32)、D87Y (n = 6)和S83F (n = 1),同时还有4个质粒介导的喹诺酮耐药基因(plasmid-mediated quinolone resistance, PMQR),分别为oqxAB (n = 47)、qnrB6 (n = 9)、qnrS2 (n = 6)和qepA (n = 1)。此外,在13株编码ESBLs的分离株中发现三种blaCTX-M亚型,分别为blaCTX-M-55(n = 9)、blaCTX-M-14(n = 3)和blaCTX-M-27(n = 1);还有8株分离株携带介导阿奇霉素耐药的mph(A)基因。在73株具有典型ASSuT耐药谱的分离株中,51株携带典型耐药基因[blaTEM-1B、strAB、sul2和tet(B)],这些耐药基因插入在fljBA操纵子区域,使上述分离株具有与欧洲分离株类似的单相表型和耐药特征的遗传背景(附录A中的表S5)[39]。值得关注的是,进化支2中有三株分离株检测到可移动黏菌素耐药基因mcr-1。
《图2》
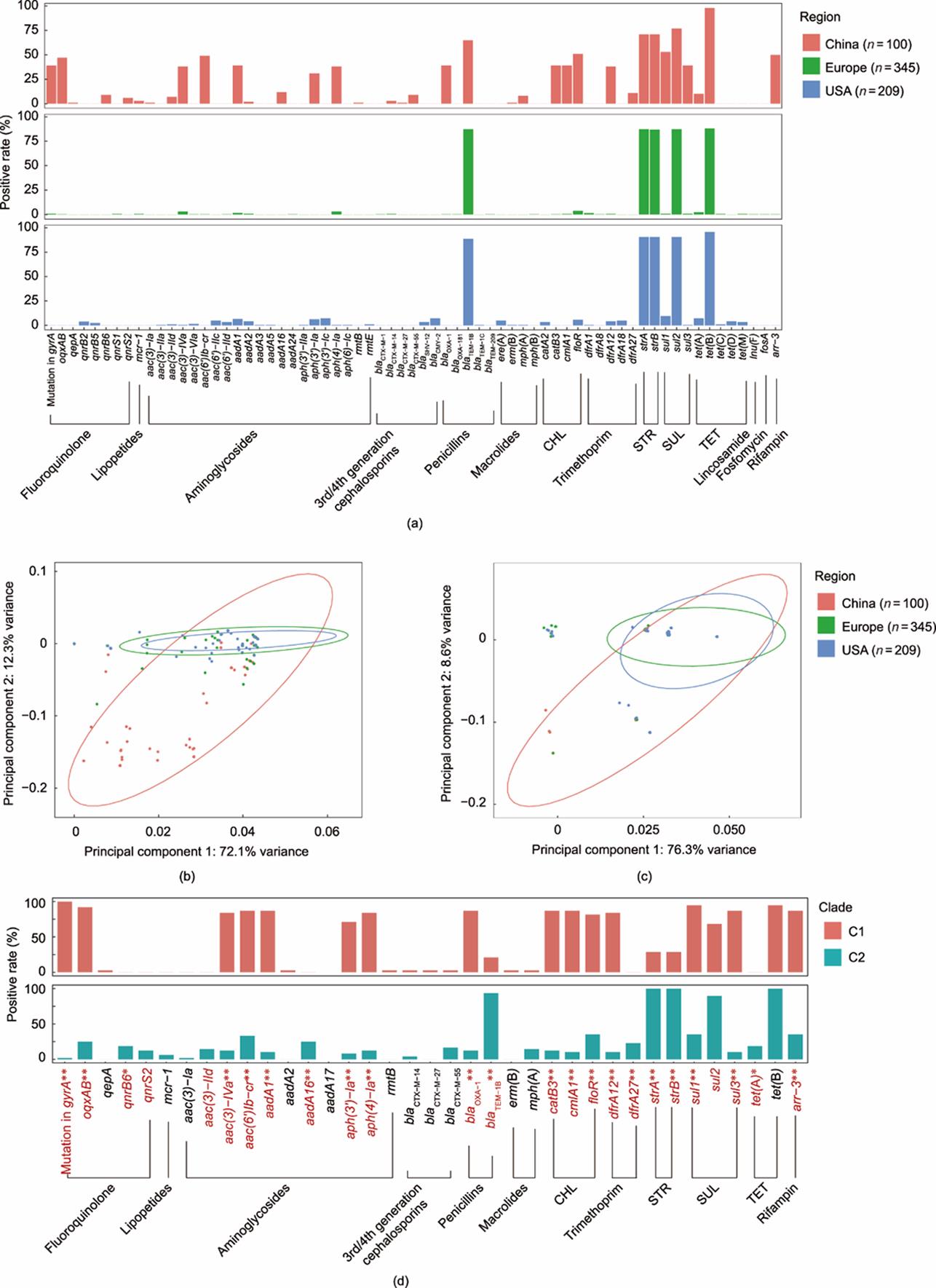
图2 (a)中国、欧洲和美国S. 4,[
《图3》
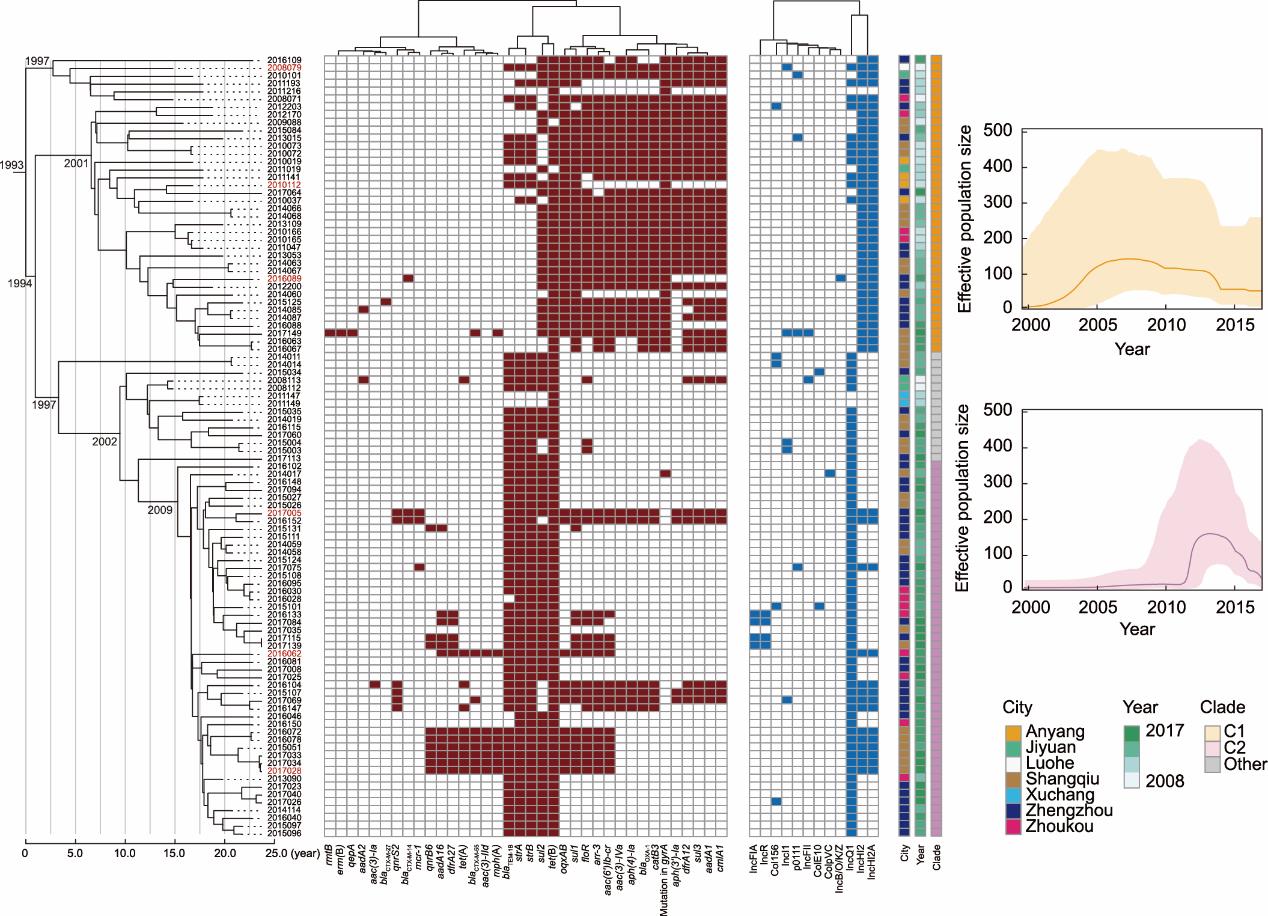
图3 中国S. 4,[
《3.3 不同地区分离株的系统发育关系及耐药基因差异》
3.3 不同地区分离株的系统发育关系及耐药基因差异
本研究收集的100株腹泻患者分离的S. 4,[5],12:i:-均为ST34型,因此从NCBI数据库下载了558株分别来自欧洲(n = 345)、美国(n = 209)和中国(n = 4)猪源的ST34 S. 4,[5],12:i:-(附录A中的表S6)一同进行全基因组比较分析。基于基因组SNP分析共获得4261个核心基因组SNP位点(菌株两两间存在0~138个差异位点,中位数为41),通过最大似然法构建了核心基因组系统发育树[图1(a)]。中国腹泻患者的分离株主要分为进化支C1 (n = 38)和C2 (n = 48)两个进化分支,在河南省广泛分布[图1(b)],其余14株散布在欧洲和美国来源菌株的遗传分支上,与C1和C2的遗传距离较远,4株中国猪源分离株位于C1分支。C1和C2分支均归属于欧洲谱系,其起源可能来自欧洲谱系,经历了两次不同的进化事件形成了目前的两个流行株系,进化支C2于2013年出现,2017年在所有S. 4,[5],12:i:-分离株中的比例增至80% (16/20) [图1(c)]。
C1分离株gyrA基因突变、PMQR基因以及抗氨基糖苷类、氯霉素、甲氧苄啶和利福平耐药基因的携带率普遍高于欧洲及美国来源菌株[图2(a)、(d)]。欧洲和美国来源菌株的耐药基因分布和质粒类型相似,但和中国腹泻患者分离株有明显不同[图2(b)、(c)]。以上结果提示中国腹泻患者分离株可能通过获得新的质粒和染色体突变具有更多的耐药相关元件。
《3.4 携带不同耐药基因的不同中国进化支的演化过程》
3.4 携带不同耐药基因的不同中国进化支的演化过程
系统发育关系、携带耐药基因和质粒类型的特征表明,大多数中国分离株起源于欧洲,在进化过程中获得了携带耐药基因的新质粒。本研究进一步分析了中国分离株的系统进化过程及质粒、耐药基因的演变规律。通过贝叶斯分析推断了中国分离株的分化节点,100株中国腹泻患者分离株的最早共同祖先出现于1993年(95%置信区间:1987—1998年),C2分离株出现于2009年(95%置信区间:2005—2011年)(图3)。通过估计有效种群规模随时间推移的变化轨迹,发现C1和C2的遗传多样性变化特征存在差异(图3)。C1分支在21世纪初首次出现后有效种群规模开始增长,自2010年后下降。C2分支呈相反趋势,2010年以后有效种群规模快速上升,这与同时期观察到的C2分离株所占比例的快速增加一致[图1(c)]。
C1和C2分离株的耐药基因分布有显著差异(图3)。C1分离株对氟喹诺酮(由gyrA基因点突变和oqxAB基因介导)、氨基糖苷类[aadA1、aph(3´)-Ia、aph(4)-Ia、aac(6´)Ib-cr和aac(3)-Iva]、氯霉素(catB3、cmlA1和floR)和利福平(arr-3)的耐药率显著高于C2(p < 0.05)。C2分离株的blaTEM-1B(氨苄西林耐药)和strAB(链霉素耐药)的携带率显著高于C1 [p < 0.05,图2(d),附录A中的表S7]。对青霉素、甲氧苄啶和磺胺类药物的耐药基因分析表明,各进化支的常见耐药基因有所区别:blaOXA-1、dfrA12、sul1和sul3主要存在于C1,而blaTEM-1B和strAB在C2中更常见。此外,欧洲、美国和中国来源的所有分离株均携带沙门菌基因岛4(Salmonella Genomic Island-4, SGI-4),该毒力岛是编码与银、砷和铜等重金属耐受相关基因的80 kbp的可移动遗传元件,可能是S. 4,[5],12:i:-典型且保守的遗传元件。聚类在C1和C2以外的14株中国分离株,其耐药基因和质粒的携带特征与C2分离株相似(图3)。
《3.5 耐药相关质粒的多态性》
3.5 耐药相关质粒的多态性
C1和C2分支携带的质粒种类不同,C2中主要为IncQ1质粒(复制子),C1主要为IncHI2和IncHI2A质粒(图3)。与鼠伤寒沙门菌LT2的比较基因组学分析表明,IncQ1复制子通常与ASSuT耐药基因[blaTEM-1B、strAB、sul2和tet(B)]一起插入染色体fljBA操纵子。携带InHI2和InHI2A质粒分离株的耐药基因谱因其来源而异[图4(a)]。美国分离株携带tet(D)、dfrA18、blaSHV-12和catA2等9种耐药基因,中国来源的C2进化支还携带另外7种耐药基因[qnrB6、dfrA27、blaCTX-M-55和mph(A)等]。本研究中,14种耐药基因分布与IncHI2和InHI2A质粒高度相关,而blaTEM-1B、strAB、sul2和tet(B)的分布与IncQ1复制子高度相关,aph(3´)-Ic、tet(A)、tet(M)、aadA2和blaCMY-2与IncA/C2高度相关[图4(b)]。
《图4》
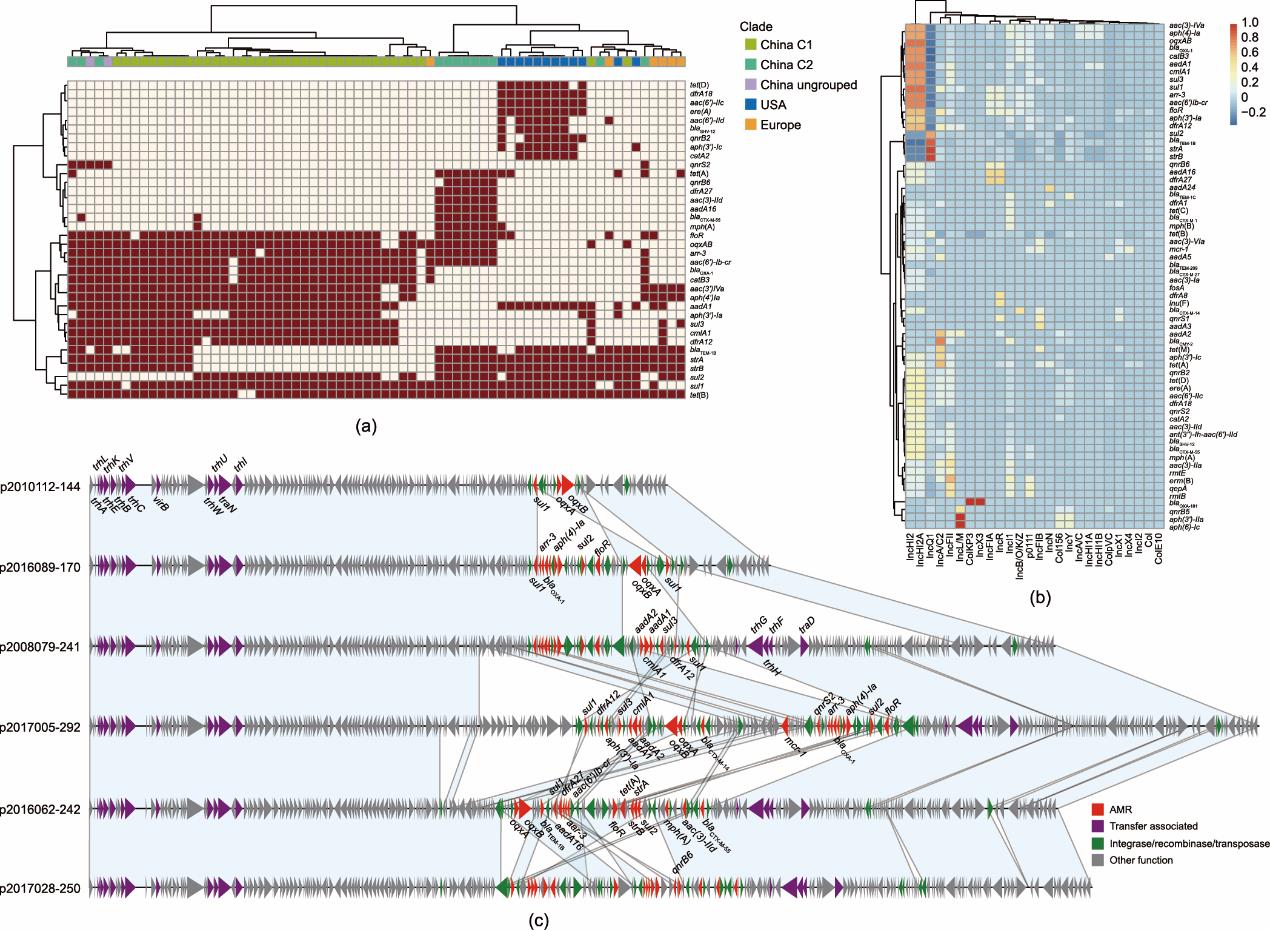
图4 (a)不同进化支中携带IncHI2-IncHI2A质粒分离株中耐药基因的分布情况。热图中红色方块表示存在该耐药基因,在少于5株分离株中存在的耐药基因未显示。(b)本研究中获得的100株中国分离株、获得基因组数据的欧洲和美国分离株中耐药基因和质粒型的相关分析结果,热图显示了相关系数。(c)6株中国代表株中多耐药IncHI2质粒的线性比对结果。浅蓝色块表示序列间的匹配区域,不同颜色的箭头表示与耐药性和移动性相关的基因(红色表示耐药基因,绿色表示整合酶、重组酶和转座子相关基因,紫色表示接合相关基因,灰色表示其他基因)。
为进一步探究耐药相关质粒的遗传背景,对中国C1和C2进化支的6株代表性菌株[图3、图4(c)]进行了三代长读长测序并获得了完整环化的染色体及质粒序列(表1)。6株分离株分别携带包含IncHI2-IncHI2A质粒在内的1~3个质粒,编码多个耐药基因。IncHI2质粒的宿主分布广泛,可介导抗生素耐药基因的传播,常与其他类型质粒发生重组[40‒41]。共鉴定4种IncHI2-IncHI2A质粒(p2010112-144、p2016089-170、p2008079-241和p2017005-292)和两种IncHI2-IncHI2A-IncQ1杂合质粒(p2016062-242和p2017028-250)。4种IncHI2-IncHI2A质粒大小为144~292 kbp(表1),由于多重耐药编码区域的可塑性,其分子结构和耐药基因有所差异,其高可塑性使宿主菌株更易获取多种耐药基因[42]。例如,mcr-1和铜抗性基因簇cusCFBA存在于质粒p2017005-292但未在其他三种质粒中检出。质粒p2017005-292序列与NCBI收录的两个鼠伤寒沙门菌中的IncHI2-IncHI2A质粒pSH16G4498(MH522423)和pS585_1(CP061116)高度同源[图5(a)]。有趣的是,中国腹泻患者分离株2008079、2010112、2016062、2017005和2017028的染色体上携带IncQ1复制子。编码IncQ1复制子及多个IS26元件的多重耐药编码区域插入染色体fljBA操纵子,导致二相鞭毛抗原编码基因失活[8]。此外,IncQ1复制子同时存在于菌株2016062和2017028的染色体和质粒上,提示携带该复制子的多重耐药编码区域可能通过插入序列(如IS26)介导进行同源重组,该假设还需要进一步验证。
《表1》
表1 使用纳米孔长读长测序获得基因组完成图的6株代表性 4,[],12:i:-菌株的基本信息和基因组特征
| Isolate | Region | Clade | Contigs (accession numbers) | Resistance genes |
|---|---|---|---|---|
| 2008079 | Luohe | C1 | 2008079-chromosome (CP090529) p2008079-241 (CP090530) 2008079-p2 (CP090531) 2008079-p3 (CP090532) | |
| 2010112 | Anyang | C1 | 2010112-chromosome (CP090541) p2010112-144 (CP090542) 2010112-p2 (CP090543) 2010112-p3 (CP090544) | |
| 2016089 | Zhengzhou | C1 | 2016089-chromosome (CP090535) p2016089-170 (CP090536) 2016089-p2 (CP090537) 2016089-p3 (CP090538) | |
| 2016062 | Zhoukou | C2 | 2016062-chromosome (CP090539) p2016062-242 (CP090540) | |
| 2017005 | Zhengzhou | C2 | 2017005-chromosome (CP090533) p2017005-292 (CP090534) | |
| 2017028 | Shangqiu | C2 | 2017028-chromosome (CP090545) p2017028-250 (CP090546) |
《图5》

图5 (a)本研究中获得的IncHI2-IncHI2A质粒以及在NCBI在线比对获得的相似质粒的序列比对结果;(b)本研究中获得的IncHI2-IncHI2A-IncQ1质粒以及在NCBI在线比对获得的相似质粒的序列比对结果。最外侧的红色环表示比对中使用的参考质粒。
两个IncHI2-IncHI2A-IncQ1质粒大小分别为242 kbp和250 kbp,具有相似的质粒骨架结构,与NCBI收录的大肠埃希菌质粒pE105-4(CP072315)和pEC22-CTX-M-15(CP084901)相似[图5(b)]。这些IncHI2-IncHI2A-IncQ1质粒携带14~18个耐药基因,包括floR、blaCTX-M-55、tet(A)和sul1。通过NCBI在线BLASTn分析表明,IncHI2-IncHI2A和IncQ1在细菌中共存现象很常见,推测在鼠伤寒沙门菌中,IncQ1通过移动元件插入染色体fljBA操纵子导致单相表型,形成沙门菌抗原式S. 4,[5],12:i:-。
《3.6 与. 4,[],12:i:-血清型单相变异相关的染色体变异》
3.6 与. 4,[],12:i:-血清型单相变异相关的染色体变异
基于6株中国腹泻患者来源的分离株的基因组完成图,我们分析了影响fljBA操纵子序列结构与血清型单相变异相关的染色体变异遗传基础。除分离株2016089外,其余5株分离株的插入序列基本一致,包括blaTEM-1B、strAB、sul2和tet(B)耐药基因和两侧的IS26序列,介导菌株ASSuT耐药表型。分离株2008079的序列较为特殊,该菌株的插入序列中blaTEM-1B相邻区域存在hin和fliC的序列片段。此外序列的插入位点有所差异,在分离株2008079和2010112中,片段插入fljB操纵子导致hin和部分fljB缺失[图6(a)];在分离株2016062、2017005和2017028中,该片段以反向插入染色体,与鼠伤寒沙门菌LT2相比导致出现STM2745到hin基因的大段缺失[图6(b)]。该插入序列与S. 4,[5],12:i:-菌株105/7/03中发现的片段高度相似;5株中国分离株的耐药区域RR1和RR2发生了重组。分离株2016089与鼠伤寒沙门菌LT2比较,分离株基因组在2 916 036~3 323 378 bp间发生约407 kbp的倒位,导致fljA和部分fljB缺失(附录A中的图S1)。
《图6》

图6 5株中国分离株中导致fljBA操纵子变异的插入序列。浅蓝色块表示序列间的匹配区域,其上的数字表示序列一致性,不同颜色的箭头表示与耐药性和移动性相关的基因(红色表示耐药基因,绿色表示整合酶、重组酶和转座子相关基因,紫色表示接合相关基因,深蓝色表示质粒复制子,灰色表示其他基因)。
《3.7 毒力基因分布》
3.7 毒力基因分布
对沙门菌毒力岛(Salmonella pathogenicity island, SPI)的携带特征进行分析表明,中国来源S. 4,[5],12:i:-携带的毒力岛高度一致,均携带SPI-1、SPI-2、SPI-3、SPI-4、SPI-5、SPI-6、SPI-9、SPI-11、SPI-12、SPI-13和SPI-14(附录A中的表S8)。国内外来源的S. 4,[5],12:i:-携带的毒力基因相似,均携带与黏附(lpf、misL、ratB、shdA和sinH)、入侵(inv)和应激(sodCI)有关的多种毒力基因,毒力基因的数量在104~108个之间(附录A中的表S9)。此外,所有纳入分析的基因组都缺少典型的鼠伤寒沙门菌毒力质粒pSLT。
《4、 讨论》
4、 讨论
S. 4,[5],12:i:-在欧洲、美国、澳大利亚和泰国的日益流行引起了全球科学家的关注[3,9,43‒44]。多重耐药ST34型S. 4,[5],12:i:-起源于欧洲并在全球广泛传播,导致动物(尤其是猪)和人类感染。中国是猪肉消费大国,且猪肉中S. 4,[5],12:i:-检出率高,因此开展中国S. 4,[5],12:i:-的分子流行病学及在国内外传播规律研究尤为重要。研究结果表明,S. 4,[5],12:i:-已超过鼠伤寒沙门菌成为河南省沙门菌感染病例中分离率排名第二的血清型。系统发育分析表明中国来源分离株分为两个主要进化支C1和C2,均归属于欧洲谱系。贝叶斯分析表明中国进化支起源于欧洲谱系,于2000年前后演变为独特的中国谱系。此外,美国谱系也起源于欧洲谱系,于21世纪初进化为美国谱系[45]。欧洲谱系可能是全球S. 4,[5],12:i:-的起源。
IS26介导的序列插入破坏了fljBA操纵子,抑制二相鞭毛抗原表达,形成血清型S. 4,[5],12:i:-,赋予菌株ASSuT耐药表型[blaTEM-1、strAB、sul2和tet(B)] [7,39,46]。除部分C1外,中国大部分分离株均携带耐药基因blaTEM-1和strAB,与IncQ1复制子高度相关,形成耐药区域RR1插入到fljBA操纵子使其失活[7]。IncQ1复制子同时存在于2016062和2017028等多个分离株的染色体RR1区域和IncHI2质粒上,表明RR1可能通过IS26介导的重组由IncHI2质粒而来。
基因组分析表明,中国分离株的耐药基因多与IncHI2复制子共存,IncHI2质粒可能是耐药基因传播的主要载体。6个IncHI2质粒完整序列比对结果表明,质粒含有一个由不同移动元件和多种耐药基因组成的多重耐药区域。IncHI2质粒是多种重要耐药基因blaCTX-M、oqxAB、qnrB和mcr的转移载体,这些耐药基因广泛存在于肠杆菌属中[46‒51]。最近报道显示IncHI2质粒可与其他类型质粒发生共整合[41]。多重耐药IncHI2质粒在S. 4,[5],12:i:-中的流行对该血清型的耐药基因种类及外源基因获取起关键作用[47]。
中国来源分离株均具有相似的毒力基因,表明该染色体区域较保守,SPI在中国、美国和欧洲来源分离株中的分布也基本一致。此外,SGI-4是典型的整合性接合元件,与铜耐受相关[52]。SGI-4可能在传播过程容易缺失,因此猪饲料中的金属补充剂的持续选择压力对SGI-4的保留至关重要[3,52‒53]。中国来源分离株中SGI-4的高流行率与欧洲分离株一致,表明其与猪肉或猪来源菌株密切相关。此外,耐药S. 4,[5],12:i:-相较鼠伤寒沙门菌能有效感染宿主的吞噬细胞和上皮细胞,但不引发显著的细胞毒性[9],其潜在的分子机制尚待深入研究。
尽管本研究针对中国腹泻患者S. 4,[5],12:i:-进行了长时间段的分子流行病学分析,但仍然存在一定的局限性。全球不同地区肉猪和被污染的猪肉是人类感染S. 4,[5],12:i:-的重要来源[43‒44,54],开展人类、动物和食品来源菌株间的比较研究将更有助于掌握该血清型在中国及世界范围内的传播规律。此外,应纳入其他省份的数据,从国家层面开展更全面系统的研究。
《5、 结论》
5、 结论
本文针对ST34型S. 4,[5],12:i:-的全基因组分析首次揭示了该血清型在中国的流行特征。中国进化支可能源于欧洲谱系,进化成具有独特耐药模式、质粒及毒力岛等移动元件携带特征的不同支系。基于“同一健康”理念开展后续研究将更全面地掌握其传播规律,并有助于采取有效措施遏制ST34型S. 4,[5],12:i:-在全球的流行。











 京公网安备 11010502051620号
京公网安备 11010502051620号




