
《1、 引言》
1、 引言
心血管疾病(cardiovascular diseases, CVD)是一类涉及心脏和血管的疾病。目前,心血管疾病死亡率占所有疾病患者死亡人数的40%以上,是人类死亡的主要病因,在全球范围内造成巨大的经济负担,属于严重的公共健康问题[1‒3]。心肌梗死(myocardial infarction, MI)是最常见的心血管疾病之一。由于氧气(O2)和营养物质供应减少引起代谢严重失衡,导致微环境稳态障碍,大量细胞死亡,炎症细胞浸润,成纤维细胞活化等。心肌梗死的这些病理性改变可能会进一步诱发心脏收缩功能障碍、心脏重塑、心肌纤维化,甚至心源性猝死[4]。
长链非编码RNA(lncRNA)是一种长度超过200个核苷酸(nt)的非编码RNA [5],它们参与许多疾病的病理生理过程和生物学过程,包括基因组印记、RNA选择性剪接和染色质修饰[6]。越来越多的研究表明,lncRNA在多种心血管疾病的表达谱中存在差异,如心肌肥厚、心律失常和心脏纤维化等。Triadin(Trdn)是一种心肌细胞特异性的lncRNA,它的敲除将导致心肌钙稳态失衡,增加小鼠心律失常易感性[7]。此外,有证据表明,lncRNA参与心肌梗死的病理过程[8‒10],如lncRNA锌脂蛋白反义链1(ZFAS1)已被证明可以作为肌浆网钙ATP酶2a(SERCA2a)抑制剂,并通过与SERCA2a结合导致心肌梗死后心脏收缩功能障碍[11]。lncRNA KLF3-AS1在心肌梗死后心肌细胞焦亡中发挥潜在作用[12]。由此可见,lncRNA在调节心肌细胞死亡和心肌重塑中具有重要作用,并有可能成为未来治疗心肌梗死的新靶点。
越来越多的证据表明,心肌梗死伴随着心脏能量代谢的改变和心肌细胞的大量流失。据报道,能量和底物[如葡萄糖和脂肪酸(FA)]供应不足是心肌梗死后细胞死亡引起心肌损伤的决定性因素[13‒14]。此外,血浆和心肌中非特异性炎症指标[如白细胞介素(IL)家族]的持续上调与心肌梗死后较差的预后相关。迄今为止,一些转录因子、细胞因子、酶和生长因子已被证实与上述病理过程相关。核因子kappa B(NFκB)是一种关键的转录因子,在许多病理生理过程中调控基因表达和介导细胞质复合物的核转位[15]。据报道,QNZ(NFκB抑制剂)通过激活葡萄糖转运蛋白4(GLUT4)来提高葡萄糖摄取,并通过抑制NFκB的激活来抑制软骨细胞变性[16]。此外,NFκB还可以作为上游信号蛋白参与调控心肌梗死后心肌细胞焦亡[17]。有证据表明,NFκB在心肌梗死过程中对血管内皮细胞和心肌细胞的功能调节也起着至关重要的作用[18‒19]。
本研究旨在探究保守性lncRNA-CPAL在心肌能量代谢紊乱和炎性心肌细胞死亡中的作用及调控机制。本研究首次发现lncRNA CPAL在心肌梗死中上调,导致三磷酸腺苷(ATP)含量减少,糖脂代谢相关蛋白分化抗原簇36(CD36)、己糖激酶1(HK1)和GLUT4表达下调,促进心肌细胞代谢功能障碍,诱导细胞焦亡。进一步研究发现NFκB是连接CPAL与心肌梗死后心肌细胞的能量代谢及炎症反应的关键调节因子,NFκB被CPAL磷酸化,加速从细胞质转运至细胞核,促进含半胱氨酸的天冬氨酸蛋白水解酶1(caspase-1)前体(pro-caspase-1)的转录与激活,进而导致心肌细胞焦亡。本研究提示CPAL可作为心肌梗死后心肌细胞代谢功能障碍和心肌细胞焦亡的一种新的调节剂和治疗靶点。
《2、 材料与方法》
2、 材料与方法
《2.1 动物》
2.1 动物
健康雄性C57BL/6小鼠(20~25 g)购自哈尔滨医科大学附属第二医院实验动物中心,并在标准动物室条件下饲养[20]。AAV9-ZsGreen-shRNA-NC(AAV9-sh-NC)和AAV9-ZsGreen-shRNA-CPAL(AAV9-sh-CPAL)经小鼠尾静脉注射。四周后,用2,2,2-三溴乙醇(T48402; Sigma-Aldrich, USA)腹腔注射麻醉小鼠,并通过小型动物呼吸机(型号VFA-23-BV;Kent Scientific, USA)使小鼠进行气道机械通气,维持正常呼吸。随后进行急性心肌梗死模型手术,在胸腔第三和第四肋骨之间打开,暴露心脏,用7-0尼龙线在冠状动脉左前降支结扎(LAD),结扎后关闭胸腔[21]。术后24 h,对心脏取材做进一步分析。将雄性小鼠随机分为四组:假手术组、心肌梗死模型组、心肌梗死模型+空病毒组(+AAV9-sh-NC组)和心肌梗死模型+心脏特异性敲减CPAL组(+AAV9-sh-CPAL组)。本研究所有动物的使用均按照哈尔滨医科大学实验动物管理准则进行,并经哈尔滨医科大学伦理委员会批准。
《2.2 CPAL敲减病毒的构建》
2.2 CPAL敲减病毒的构建
构建携带靶序列(5´-AAACATTAACGAATTAAGACC-3´)和启动子CAG表达控制的增强型绿色荧光蛋白(ZsGreen)的shRNA敲减的腺相关病毒9(Adeno-associated virus 9, AAV9)载体,给予每只小鼠尾静脉注射100 uL病毒(病毒滴度为1 × 1012 vg·mL-1)[22]。
《2.3 超声心动图》
2.3 超声心动图
用2,2,2-三溴乙醇(T4802; Sigma-Aldrich)腹腔注射麻醉小鼠。使用保温垫维持小鼠基础体温,通过超声心动图评估左心室功能[4]。
《2.4 三苯基四唑氯化物(TTC)染色》
2.4 三苯基四唑氯化物(TTC)染色
取出每组小鼠心脏组织,并在-80 °C下快速冷冻,然后将心脏组织切成2 mm厚的薄片,将薄片放在1%的TTC(Sigma-Aldrich, USA)磷酸盐溶液中孵育(37 °C, 20 min)后进行拍摄。
《2.5 细胞培养、加药处理及小干扰RNA(siRNA)转染》
2.5 细胞培养、加药处理及小干扰RNA(siRNA)转染
心肌细胞来源于新生乳鼠(1~3日龄)。将新生乳鼠用75%的酒精清洗,用无菌镊子取出心脏,将心脏组织切成小块。消化后将所得细胞悬液离心,再将其铺设在含有100 μg∙mL-1链霉素、100 U∙mL-1青霉素和10%胎牛血清的细胞培养瓶中。将分离的细胞置于细胞培养箱中37 °C培养1.5 h。将心肌细胞与成纤维细胞分离,将心肌细胞放置于细胞培养箱中继续培养(37 °C, 5% CO2)。培养48 h后的心肌细胞用于后续实验[23]。在脂多糖(LPS)刺激(50 μmol∙L-1, 12 h)前,分别给予5 μg∙mL-1可渗透细胞的NFκB抑制剂SN50(HY-P015; MCE, USA)和50 μg∙mL-1 caspase-1抑制剂Ac-YVAD-CMK(178603-78-6; Cayman, USA)预处理细胞30 min。
GenePharma公司合成CPAL特异性siRNA(si-CPAL)和乱序阴性对照RNA(si-NC)。CPAL的序列:正义链5´-GGUCUUAAUUCGUUAAUGUTT-3´和反义链5´-ACAUUAACGAAUUAAGACCTT-3´。使用转染试剂Lipofectamine 2000(Invitrogen, USA)将CPAL特异性siRNA包被并转染到心肌细胞[24],最终浓度为100 nmol∙L-1。转染48 h后,用于后续实验。
《2.6 Hematoxylin-Eosin染色》
2.6 Hematoxylin-Eosin染色
取各组小鼠心脏组织,放置于4%多聚甲醛固定24 h,用石蜡包埋,然后将组织切成5 μm厚的切片;将组织切片放置于二甲苯溶液中脱蜡,然后分别放置于降梯度乙醇中水合。根据苏木精-伊红染色试剂盒(G1120; Solarbio, China)使用说明书,对处理后的组织切片进行染色,并通过显微镜(FV300; Olympus, Japan)捕获图像。
《2.7 透射电子显微镜法》
2.7 透射电子显微镜法
用0.25%胰蛋白酶消化心肌细胞(37 °C, 1 min),各组心肌细胞及心脏组织用2.5%戊二醛固定后,再置于1% OsO4溶液中固定2 h,然后用1%乙酸铀酰染色,分别放置于降梯度乙醇中水合后,嵌入环氧树脂包埋;切片后进行电子染色,置于电子显微镜(JEM-1200; JEOL Ltd., Tokyo)下观察。
《2.8 dUTP缺口端标记(TUNEL)测定》
2.8 dUTP缺口端标记(TUNEL)测定
根据TUNEL检测试剂盒(Roche, Germany)说明书进行TUNEL染色,用于测定心肌细胞中DNA片段完整性[25]。
《2.9 蛋白质印迹》
2.9 蛋白质印迹
通过BCA蛋白试剂盒(P0012; Beyotime, China)检测总蛋白含量[26]。蛋白样品通过10%~13%的十二烷基硫酸钠聚丙烯酰胺凝胶电泳分离后,转移到硝酸纤维素(NC)膜上,随后放于含有5%的脱脂奶粉中封闭。然后将膜与NLR家族pyrin域蛋白3(NLRP3)(A12694; Abclonal, China)、GSDMD/GSDMD-N(ab209845; Abcam, UK)、IL-18(A1115; Abclonal)、IL-1β(A16288; Abclonal)、caspase-1(ab207802; Abcam)、磷酸化核因子κB-P65(P-NFκB P65)(Ser536; #3036; CST, USA)、NFκB P65(#8242T; CST),CD36(74002S; CST)、GLUT4(AF5386; Affinity Biosciences, USA)和HK1(19662-1-AP; Proteintech, USA)的抗体在4 °C下孵育过夜。再用吐温-20磷酸盐缓冲液(PBST)洗涤后,将膜与荧光缀合抗兔IgG二级抗体(LI-COR, USA)在室温下孵育1 h。内参为甘油醛-3-磷酸脱氢酶(GAPDH)(TA-08,北京中杉金桥生物技术有限公司)。通过奥德赛红外成像系统(LI-COR, USA)扫描并分析。
《2.10 酶联免疫吸附试验》
2.10 酶联免疫吸附试验
根据酶联免疫吸附测定(ELISA)试剂盒检测的说明书,通过ELISA试剂盒检测血清中的IL-18(Elabscience, China)、IL-1β(Elabsicience, China)和ATP(MB-6783A; Enzyme Mark Biological, China)水平。
《2.11 定量实时聚合酶链反应(qRT-PCR)》
2.11 定量实时聚合酶链反应(qRT-PCR)
使用TRIzol试剂(Invitrogen, USA)分别提取心肌组织和心肌细胞中总RNA,使用逆转录试剂盒(Toyobo Co., Ltd., Japan),20 µL体系含1 µg模板RNA和4 µL 5 ×逆转录缓冲液、1 µL逆转录酶混合液、1 µL引物混合液、无核酸酶水逆转录试剂,逆转录成cDNA。然后使用SYBR PCR扩增混合试剂盒(Toyobo Co., Ltd., Japan)检测CPAL、NLRP3、caspase-1、IL-18、IL-1β、HK1和CD36基因。使用2-ΔΔCt算法,将转录产物用内源性GAPDH标准化[27]。引物(小鼠)的序列由Invitrogen公司合成,如附录A中表S1所示。
《2.12 免疫组织化学分析》
2.12 免疫组织化学分析
按照标准程序将心脏组织固定、脱水、包埋和切片。接下来,将切片的组织脱蜡、再水化并用过氧化氢封闭,然后与NLRP3(BA3677; BOSTER, China)、caspase-1(ab207802; Abcam)和P-NFκB(AF2006; Affinity Biosciences, USA)抗体在4 °C孵育过夜。用PBST洗涤三次,用二抗(LI-COR, USA)孵育1 h。使用二氨基联苯胺(DAB)(北京中杉金桥生物技术有限公司)对组织切片进行孵育,最后使用苏木精对细胞核进行染色。然后在显微镜下拍摄。使用ImageJ软件确定染色区域的大小[28]。
《2.13 免疫荧光》
2.13 免疫荧光
采用免疫荧光染色技术检测NLRP3、caspase-1和P-NFκB在心肌细胞中的表达和定位。荧光染色的方法基于之前的研究[20]。抗体如下:NLRP3(BA3677; Boster)、caspase-1(ab207802; Abcam)和P-NFκB(AF2006; Affinity Biosciences)。
《2.14 荧光原位杂交》
2.14 荧光原位杂交
通过荧光原位杂交(FISH)实验检测心肌细胞中CPAL的表达和定位。当培养的心肌细胞融合度达到60%~80%后,用预杂交液封闭处理,然后用0.5% Triton X-100穿透。通过PBS洗涤后固定,将心肌细胞与Cy3共轭的CPAL探针、18S探针及U6探针分别在杂交缓冲液中孵育。用柠檬酸钠(SSC)缓冲液和PBS洗涤后,用4´,6-二氨基-2-苯基吲哚(DAPI)进行细胞核染色。在共聚焦显微镜(Axio Scope A1, ZEISS, Germany)下分析荧光定位。
《2.15 染色质免疫共沉淀(ChIP)》
2.15 染色质免疫共沉淀(ChIP)
根据ChIP试剂盒(Invitrogen)使用说明书进行实验。利用聚合酶链反应(PCR)技术分析NFκB P65抗体、免疫球蛋白G(IgG)抗体与DNA免疫共沉淀。使用引物如下:NFκB结合在Caspase-1 DNA位点1(‒836 nt至‒846 nt)的上下游引物(正向5´-AAAGAAGCCAAGAGCCAGGT-3´和反向5´-AGTGGACCAAGGAATGGTTG-3´)和NFκB结合在Caspase-1 DNA位点2(‒1555 nt至‒1544 nt)的上下游引物(正向5´-TGTTTGGTTGGCTGGTTGTT-3´和反向5´-GGACCAGAAGCAGAGGTGTG-3´)。
《2.16 RNA结合蛋白免疫共沉淀(RIP)》
2.16 RNA结合蛋白免疫共沉淀(RIP)
根据MagnaRIP RNA结合蛋白免疫沉淀试剂盒(17-701; Millipore, Germany)说明书进行RIP实验操作[11]。简而言之,将心脏组织用RIP裂解缓冲液裂解。然后,将提取的总RNA与NFκB P65(622604; Biolegend, USA)抗体或对照抗体(IgG)混合并沉淀。接下来,通过蛋白酶K处理纯化沉淀的RNA。最后,通过qRT-PCR检测RNA。
《2.17 乳酸脱氢酶(LDH)释放试验》
2.17 乳酸脱氢酶(LDH)释放试验
根据LDH测定试剂盒(A020-2,南京建成生物工程研究所有限公司)检测说明书测定细胞中LDH的释放。
《2.18 统计分析》
2.18 统计分析
使用Graphpad Prism 5.0软件(GraphPad, Inc., USA)对实验数据进行统计分析。两组间比较采用t检验。多组比较采用单因素方差分析(ANOVA)。组数据以均值±标准误差(SEM)表示,p值小于0.05被认为具有统计学意义。
《3、 结果》
3、 结果
《3.1 心肌梗死后心肌重构和能量代谢的改变》
3.1 心肌梗死后心肌重构和能量代谢的改变
首先,为了揭示心肌梗死后心脏损伤的多种病理变化,通过小鼠心脏LAD结扎建立在体心肌梗死模型。超声心动图和血流动力学测量结果显示,与假手术组相比,心肌梗死模型组心脏的射血分数(EF)和缩短分数(FS)均显著降低[图1(a)]。苏木精和伊红染色结果显示,小鼠梗死边缘区心脏组织结构明显异常,包括心肌细胞肥大、组织间隙增加,并伴有炎性细胞浸润[图1(b)]。电镜图像结果显示,心肌梗死模型组小鼠梗死边缘区心肌组织的超微结构异常,表现为线粒体肿胀和细胞核固缩[图1(c)]。此外,为了探究心肌梗死小鼠缺血心肌的能量代谢变化(如ATP、葡萄糖和FA),本研究检测了ATP在缺血心肌中的表达水平。心肌梗死后,ATP作为心肌中能量的主要形式被利用。ELISA检测结果发现心肌梗死边缘区心肌组织中ATP水平降低[图1(d)]。在心肌梗死模型组中,GLUT4、HK1和CD36的mRNA和蛋白水平也明显下调[图1(e)和(f)]。这些结果表明,心肌梗死小鼠的心脏损伤程度可能与心肌代谢性改变和炎症损伤有关。
《图1》
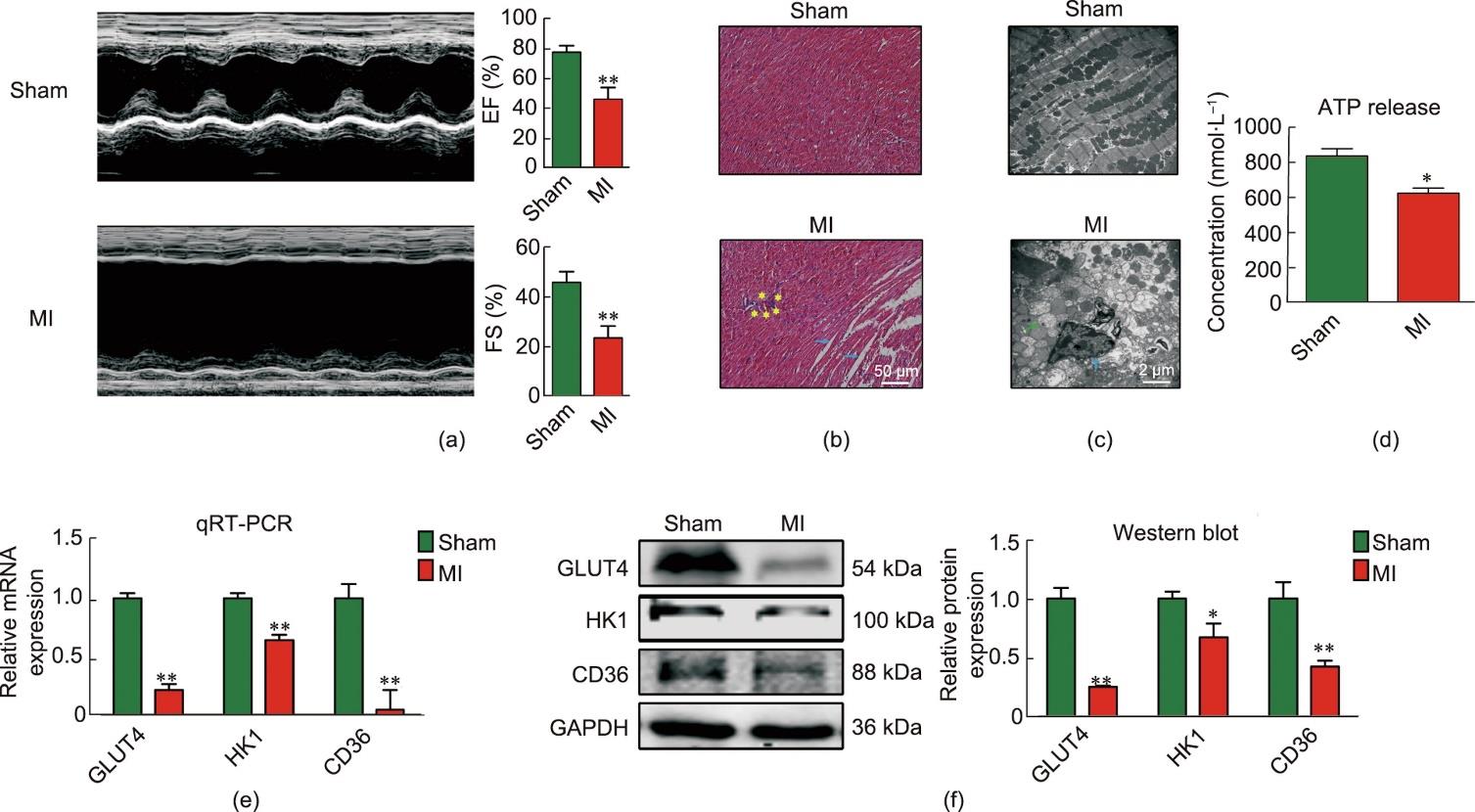
图1 心肌梗死小鼠心脏损伤评估。(a)超声心动图评估假手术组和心肌梗死组小鼠心脏EF、FS值(n = 5;**P < 0.01,与假手术组比较;均值±标准误差)。(b)假手术组和心肌梗死组小鼠心脏横切面H&E染色(n = 3;标尺:50 μm)。蓝色箭头表示细胞间隙较大,黄色星形表示炎症细胞浸润。(c)假手术组和心肌梗死组小鼠心脏代表性透射电镜(TEM)显微图;蓝色箭头表示核固缩,绿色箭头表示线粒体肿胀(n = 4;标尺:2 μm)。(d)假手术和心肌梗死组小鼠心脏组织匀浆中ATP表达水平(n = 3;* P < 0.05)。(e)、(f)心肌梗死24 h后各组小鼠左心室梗死边缘区GLUT4、HK1、CD36 mRNA和蛋白表达水平(*P < 0.05,**P < 0.01,与假手术组比较;n = 3~4;均值±标准误差)。
《3.2 心肌梗死后发生心肌细胞焦亡》
3.2 心肌梗死后发生心肌细胞焦亡
越来越多的证据表明,焦亡在心肌梗死后的炎症过程中起着关键作用[29‒30]。在典型的焦亡信号通路中,pro-caspase-1蛋白的寡聚作用引导自我蛋白酶解为caspase-1的裂解体,即活化的含半胱氨酸的天冬氨酸蛋白水解酶1(cleaved caspase-1),然后激活GSDMD的裂解并诱导GSDMD-N端片段的释放,导致膜孔释放具有生物活性的IL-18和IL-1β [28]。为了进一步探究心肌梗死后炎性细胞死亡,本研究进行TUNEL染色分析以确定心肌梗死小鼠心脏组织中DNA片段的完整性,结果显示,心肌梗死小鼠的梗死边缘区心肌组织中出现大量TUNEL染色阳性细胞[图2(a)]。qRT-PCR结果显示,在小鼠梗死边缘区心肌组织中,NLRP3、caspase-1、IL-18和IL-1β的mRNA表达水平显著增加[图2(b)]。NLRP3、cleaved caspase-1、GSDMD、GSDMD-N、前体白细胞介素-18(pro-IL-18)、成熟体白细胞介素-18(mature IL-18)、前体白细胞介素-1β(pro-IL-1β)和成熟体白胞介素-1β(mature IL-1β)的蛋白表达水平在小鼠梗死边缘区心肌组织中也显著上调[图2(c)]。免疫组化分析结果显示,在心肌梗死模型组中,NLRP3和caspase-1的表达水平显著上调[图2(d)]。此外,ELISA结果显示,心肌梗死模型组小鼠血清中IL-18和IL-1β也出现升高的类似变化[图2(e)],表明心肌梗死后出现心肌细胞焦亡现象。
《图2》
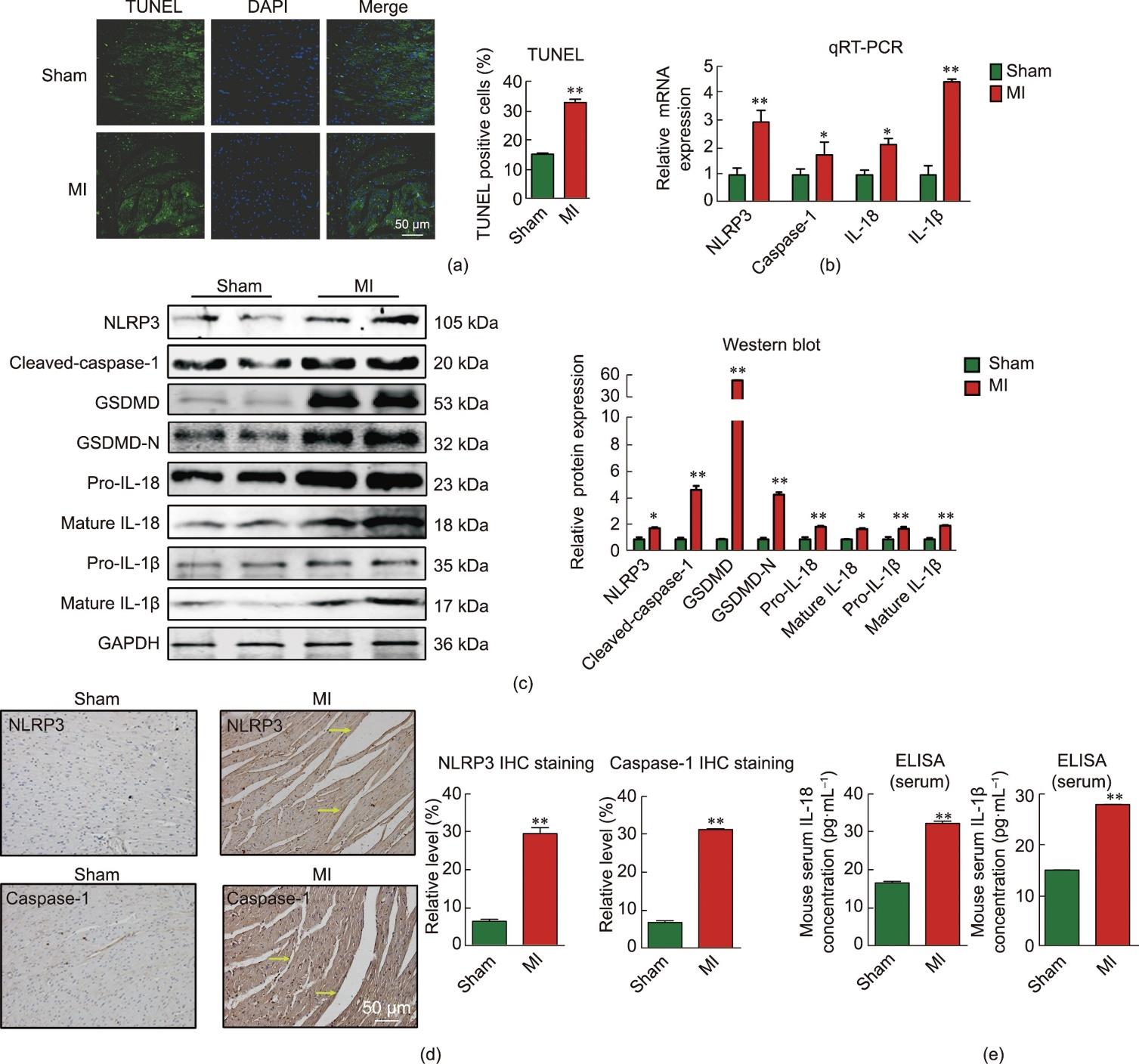
图2 心肌梗死心脏发生心肌细胞焦亡。(a)TUNEL染色显示焦亡代表性图像,即假手术组和心肌梗死组染色体DNA断裂的细胞(绿色:TUNEL阳性细胞;蓝色:DAPI;n = 3;标尺:50 μm)。(b)假手术组和心肌梗死组小鼠心脏mRNA相对表达水平(n = 4~8;*P < 0.05,**P < 0.01,与假手术组比较;均值±标准误差)。(c)假手术与心肌梗死组小鼠NLRP3、cleaved caspase-1、GSDMD、GSDMD-N、pro-IL-18、mature IL-18、pro-IL-1β和mature IL-1β蛋白相对表达水平(*P < 0.05,**P < 0.01,与假手术组比较;n = 3~9;均值±标准误差)。(d)假手术组和心肌梗死组小鼠心脏免疫组化染色显示NLRP3和caspase-1蛋白表达水平(黄色箭头代表阳性染色细胞;**P < 0.01,与假手术组比较;n = 3,均值±标准误差;标尺:50 μm)。(e)ELISA试剂盒检测假手术组和心肌梗死组小鼠血清中IL-18、IL-1β 浓度(**P < 0.01;n = 4~6,与假手术组比较;均值±标准误差)。
使用LPS诱导心肌细胞的炎症反应,以模拟心肌细胞的炎症环境。TUNEL染色结果显示LPS可诱导心肌细胞核内染色质DNA断裂[见附录A中的图S1(a)]。电镜结果显示,LPS处理的心肌细胞表现为细胞核固缩、线粒体肿胀和细胞膜溶解破裂[见附录A中的图S1(b)]。qRT-PCR分析结果显示,在LPS处理组中,caspase-1的mRNA表达水平显著增加,NLRP3、IL-18和IL-1β的mRNA表达水平也显著增加[见附录A中的图S1(c)]。同时,NLRP3、cleaved caspase-1、GSDMD、GSDMD-N、pro-IL-18、mature IL-18、pro-IL-1β和mature IL-1β的蛋白表达水平在LPS处理的心肌细胞中显著上调[见附录A图S1(d)]。免疫荧光结果显示,LPS处理使得心肌细胞中NLRP3和caspase-1的荧光强度显著增强[见附录A中的图S1(e)]。
《3.3 CPAL缺失抑制心肌梗死后心脏损伤》
3.3 CPAL缺失抑制心肌梗死后心脏损伤
接下来,探究lncRNA在心肌梗死后炎症反应过程中的关键作用。首先检测了可能与心血管疾病相关的lncRNA差异表达水平[10],其中包括AK009126(CPAL)、AK157022、AK086435、AK156356、AK076765、AK035575、AK044386、AK141702、AK0158845、ENSMUST00000148132、ENSMUST00000101162、ENSMUST00000137198、ENSMUST00000143898在假手术组和小鼠梗死边缘区心肌组织或LPS诱导的炎性心肌细胞中的表达变化。结果显示,CPAL在小鼠梗死边缘区心肌组织和LPS诱导的炎性心肌细胞中均显著上调[图3(a)和(b)]。FISH结果显示CPAL在心肌细胞的细胞核和细胞质中均有表达,并且在LPS处理的新生乳鼠心肌细胞中表达增加[图3(c)]。为了进一步探讨CPAL是否与心肌梗死后心肌细胞代谢异常和细胞焦亡相关,本研究绘制了如下的实验流程图[图3(d)]。在体水平,通过尾静脉注射的方式将AAV9-ZsGreen-shRNA-NC(AAV9-sh-NC)和AAV9-ZsGreen-shRNA-CPAL(AAV9-sh-CPAL)输送到小鼠体内,然后在CPAL敲减四周后,通过qRT-PCR实验验证CPAL 在各组织中的感染效率。结果表明,CPAL在心肌组织中被成功敲减[见附录A中的图S2]。随后,通过结扎小鼠心脏冠状动脉左前降支24 h,构建心肌梗死小鼠模型。2,3,5-TTC染色结果显示,敲除CPAL可减少心肌梗死组小鼠心脏缺血面积[图3(e)]。超声心动图结果显示,心肌梗死模型组小鼠心脏的左心室射血分数和左心室缩短分数在小鼠心肌缺血24 h后显著下降;而与心肌梗死模型组相比,CPAL敲减组小鼠的左心室射血分数和左心室缩短分数明显增加,表明敲减CPAL对心脏具有保护作用[图3(f)]。苏木精和伊红染色结果显示假手术组小鼠的心脏未出现组织学损伤。 心肌梗死模型组小鼠梗死边缘区的心肌组织表现出轻度炎症病灶,并且这些改变在CPAL敲减组中显著减少[图3(g)]。此外,为了探究CPAL对心肌细胞超微结构的作用,本研究进行了电镜检测。结果显示在心肌梗死模型组小鼠梗死边缘区的心肌组织中,心肌细胞的超微结构表现为线粒体肿胀和细胞核固缩,而CPAL敲减组中这些异常改变得到显著缓解,表明敲减CPAL可以抑制心肌梗死后炎症进程,减少心肌缺血性损伤[图3(h)]。
《图3》
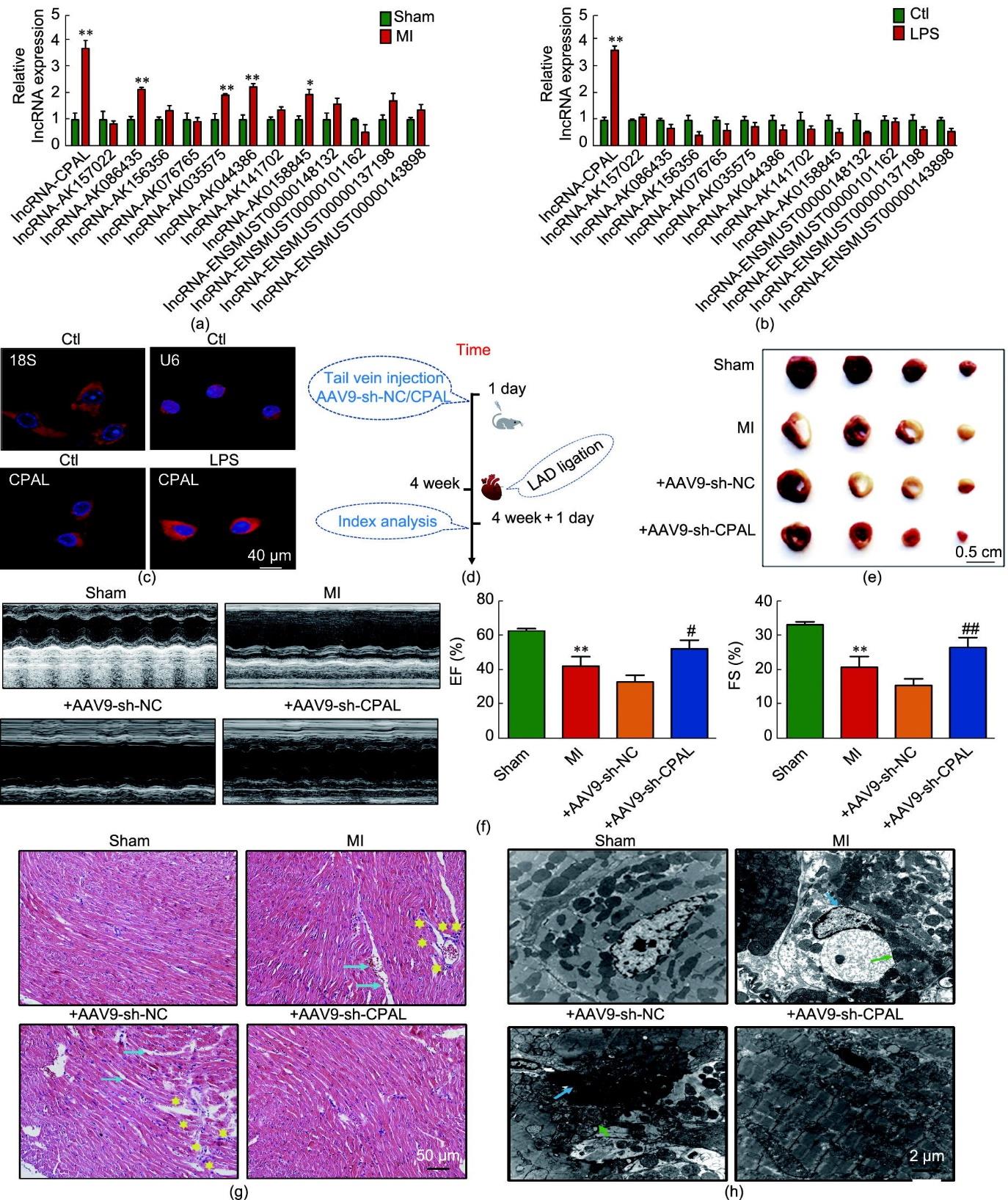
图3 CPAL在心肌梗死的心脏组中表达上调。(a)qRT-PCR实验分析假手术组与心肌梗死组小鼠心脏lncRNA表达情况(n = 3~6;*P < 0.05,**P < 0.01,与假手术组比较)。(b)验证LPS(50 nmol·L-1)处理心肌细胞12 h或未处理心肌细胞后lncRNA表达水平(n = 3~6;**P < 0.01与对照组比较)。(c)心肌细胞中CPAL特异性探针FISH染色(18S为细胞质标记物,U6为细胞核标记物;标尺:40 μm;n = 3~5)。(d)在体研究实验流程示意图。(e)携带shRNA的重组腺相关病毒(AAV9-sh-CPAL)敲减小鼠内源性CPAL。假手术组、心肌梗死组、+AAV9-shNC组、+AAV9-sh-CPAL组小鼠心脏梗死边缘区TTC染色(标尺:0.5 cm)。(f)超声心动图评估假手术组、心肌梗死组、+AAV9-sh-NC组、+AAV9-sh-CPAL组小鼠心脏EF、FS指标(**P < 0.01,与假手术组比较;#P < 0.05,##P < 0.01,与+AAV9-sh-NC组比较;n = 5~8;均值±标准误差)。(g)假手术组、心肌梗死组、+AAV9-sh-NC组和+AAV9-sh-CPAL组心脏横切面H&E染色(蓝色箭头表示细胞间隙较大,黄色星形表示炎症细胞浸润;标尺:50 μm;n = 3~4)。(h)电镜图显示各组小鼠心肌超微结构变化(蓝色箭头:核固缩;绿色箭头:线粒体肿胀;标尺:2 μm;n = 4)。
《3.4 CPAL缺失对心肌梗死后心肌细胞代谢的调控》
3.4 CPAL缺失对心肌梗死后心肌细胞代谢的调控
心肌梗死后心室重构是一个缓慢而复杂的过程,其能量代谢受多种因素的调控。本文首先探讨了CPAL是否参与NFκB相关的心肌能量代谢过程。通过ELISA检测发现,心肌梗死模型组小鼠梗死边缘区的心肌组织中ATP水平降低,然而,敲除CPAL可以明显抑制ATP的减少[图4(a)]。此外,心肌缺血时表现出糖脂代谢的异常,结果显示,HK1和CD36的mRNA表达水平在心肌梗死模型组中明显下调,而CPAL缺失可抑制其表达下调[图4(b)和(c)]。同时,GLUT4、HK1和CD36的蛋白表达水平在小鼠梗死边缘区的心肌组织中显著下调,CPAL缺失可抑制其蛋白表达下调[图4(d)~(f)]。综上所述,这些数据表明CPAL的缺失有助于减少缺血的心肌细胞发生异常的糖脂代谢,从而有助于减轻心肌缺血性损伤。
《图4》
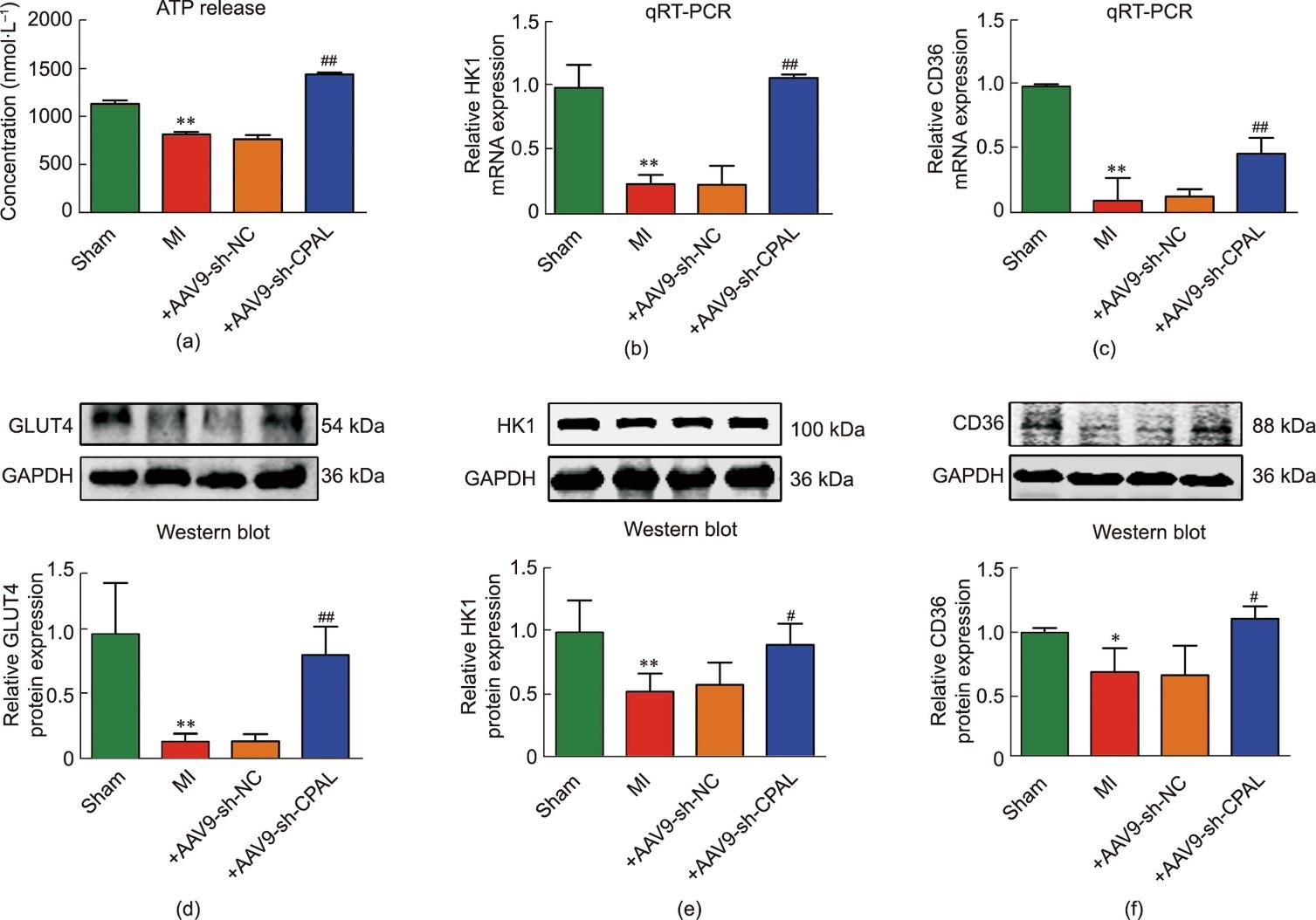
图4 CPAL调节心肌梗死小鼠心肌代谢。(a)假手术组、心肌梗死组、+AAV9-sh-NC组和+AAV9-sh-CPAL组心脏组织匀浆中ATP表达水平(n = 5;** p < 0.01,与假手术组比较)。(b)、(c)心肌梗死后24 h各组小鼠左心室梗死边缘区HK1、CD36的mRNA表达水平(**P < 0.01,与假手术组比较;##P < 0.01,与+AAV9-sh-NC组比较;n = 3~5;均值±标准误差)。(d)~(f)各组小鼠左心室梗死边缘区GLUT4、HK1、CD36相对蛋白表达水平。GAPDH作为内参进行归一化(*P < 0.05,**P < 0.01,与假手术组比较;#P < 0.05,##P < 0.01,与+AAV9-sh-NC组比较;n = 3;均值±标准误差)。
《3.5 敲除CPAL减少小鼠心肌梗死后心肌细胞焦亡》
3.5 敲除CPAL减少小鼠心肌梗死后心肌细胞焦亡
为了探究CPAL缺失是否会减少心肌梗死后心肌细胞焦亡,本文进行了TUNEL染色实验,评估心肌缺血引起的细胞死亡情况。结果显示CPAL缺失可抑制细胞死亡[图5(a)]。从图5(b)中发现,与心肌梗死模型组相比,CPAL缺失可降低NLRP3、caspase-1、IL-18和IL-1β的mRNA表达水平。与此相一致的是,蛋白质印迹分析结果显示NLRP3、cleaved caspase-1、GSDMDGSDMD-N、pro-IL-18、mature IL-18、pro-IL-1β和mature IL-1β的蛋白表达水平也因CPAL敲减而显著降低[图5(c)]。收集各组小鼠血清样本,利用ELISA试剂盒检测小鼠血清中IL-18和IL-1β的水平,结果显示,相比于心肌梗死模型组小鼠,CPAL敲减组小鼠血清中IL-18和IL-1β的水平明显降低[图5(d)]。同样,免疫组化分析结果显示,与心肌梗死模型组小鼠相比,CPAL敲减组小鼠梗死边缘区心肌组织中NLRP3和caspase-1的表达水平显著降低[图5(e)]。
《图5》
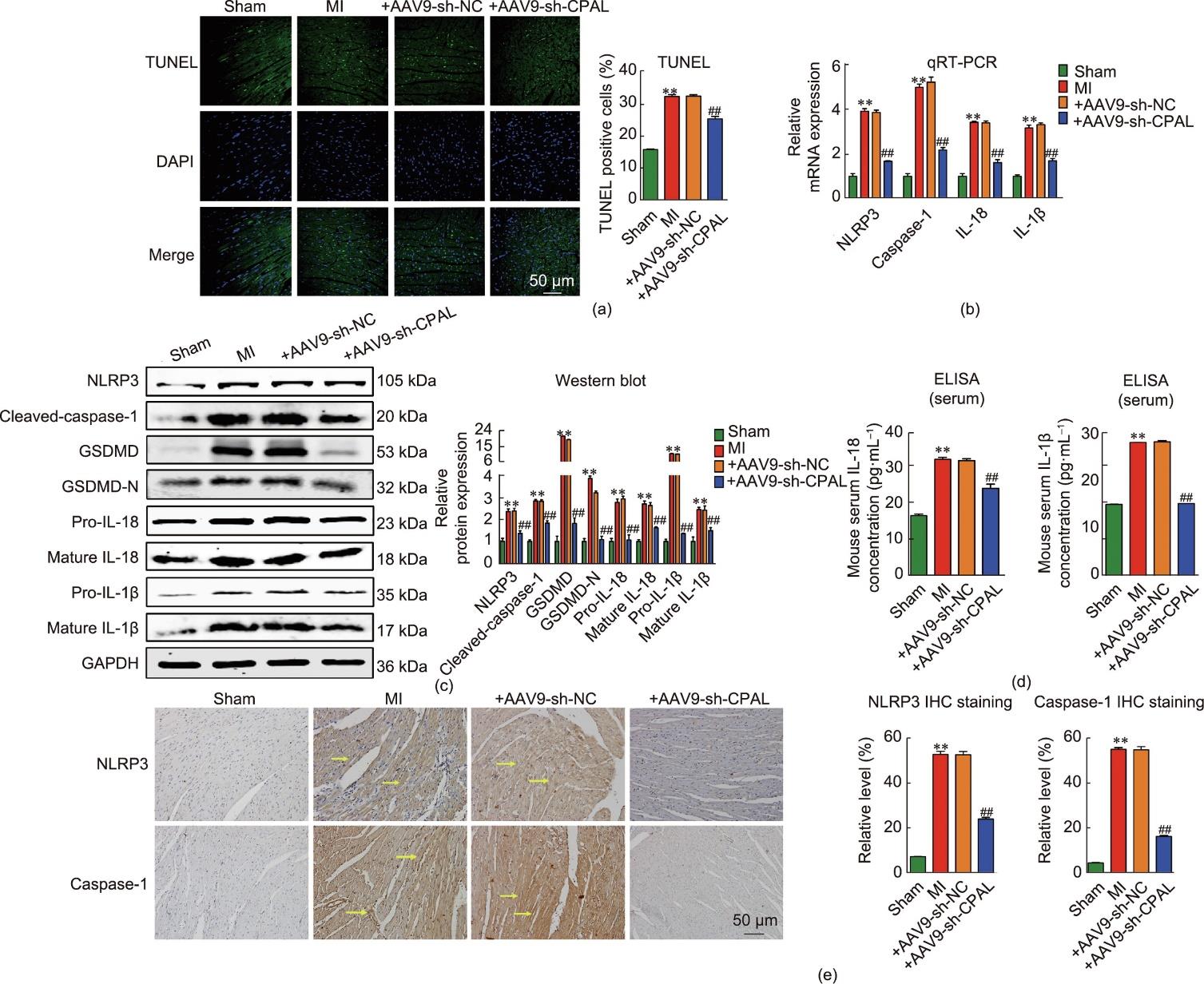
图5 CPAL下调抑制心肌梗死后细胞焦亡。(a)代表性TUNEL图像显示各组中染色体DNA断裂的细胞(绿色:TUNEL阳性细胞;蓝色:DAPI;n = 3~4;标尺:50 μm;**P < 0.01,与假手术组比较;##P < 0.01,与+AAV9-sh-NC组比较;均值±标准误差)。(b)心肌梗死术后24 h各组小鼠心脏左心室梗死边缘区NLRP3、caspase-1、IL-18、IL-1β mRNA表达水平(**P < 0.01,与假手术组比较;##P < 0.01,与+AAV9-sh-NC组比较;均值±标准误差;每组n = 6~10)。(c)小鼠心脏中NLRP3、cleaved caspase-1、GSDMD、GSDMD-N、pro-IL-18、mature IL-18、pro-IL-1β和mature IL-1β蛋白相对表达水平。GAPDH作为内参进行归一化(**P < 0.01,与假手术组比较;##P < 0.01,与+AAV9-sh-NC组比较;n = 3~6;均值±标准误差)。(d)各组小鼠血清中IL-18、IL-1β浓度(**P < 0.01,与假手术组比较;##P < 0.01,与+AAV9-sh-NC组比较;均值±标准误差;每组n = 4~6)。(e)左心室梗死边缘区NLRP3、caspase-1蛋白免疫组化染色(**P < 0.01,与假手术组比较;##P < 0.01,与+AAV9-sh-NC组比较;n = 3~4;均值±标准误差)。
《3.6 离体水平,CPAL沉默可抑制LPA诱导的心肌细胞焦亡》
3.6 离体水平,CPAL沉默可抑制LPA诱导的心肌细胞焦亡
培养小鼠原代心肌细胞,使用CPAL小干扰RNA(si-CPAL)成功沉默CPAL(见附录A中的图S3)。TUNEL染色结果显示,LPS诱导心肌细胞死亡,而沉默CPAL可显著提升心肌细胞的存活率[图6(a)]。如图6(b)所示,LPS处理组心肌细胞表现出不同程度的超微结构变化,主要包括细胞核固缩、线粒体肿胀变性和细胞膜破损,而si-CPAL组小鼠心肌细胞的这些现象明显减轻。此外,蛋白质印迹分析结果显示,与LPS + si-NC处理组相比,沉默CPAL后,NLRP3、cleaved caspase-1、GSDMD、GSDMD-N、pro-IL-18、mature IL-18、pro-IL-1β和mature IL-1β的蛋白表达水平显著下调[图6(c)]。与之类似,免疫荧光结果显示,与+si-NC组相比,+si-CPAL组的小鼠心肌细胞中NLRP3和caspase-1的荧光强度显著降低[图6(d)]。此外,分析了CPAL对LDH释放的影响,结果显示,沉默CPAL后可以明显抑制由LPS刺激导致的心肌细胞LDH释放水平的增加。这些结果表明质膜通透性的增加与CPAL介导的心肌细胞焦亡相关[图6(e)]。
《图6》
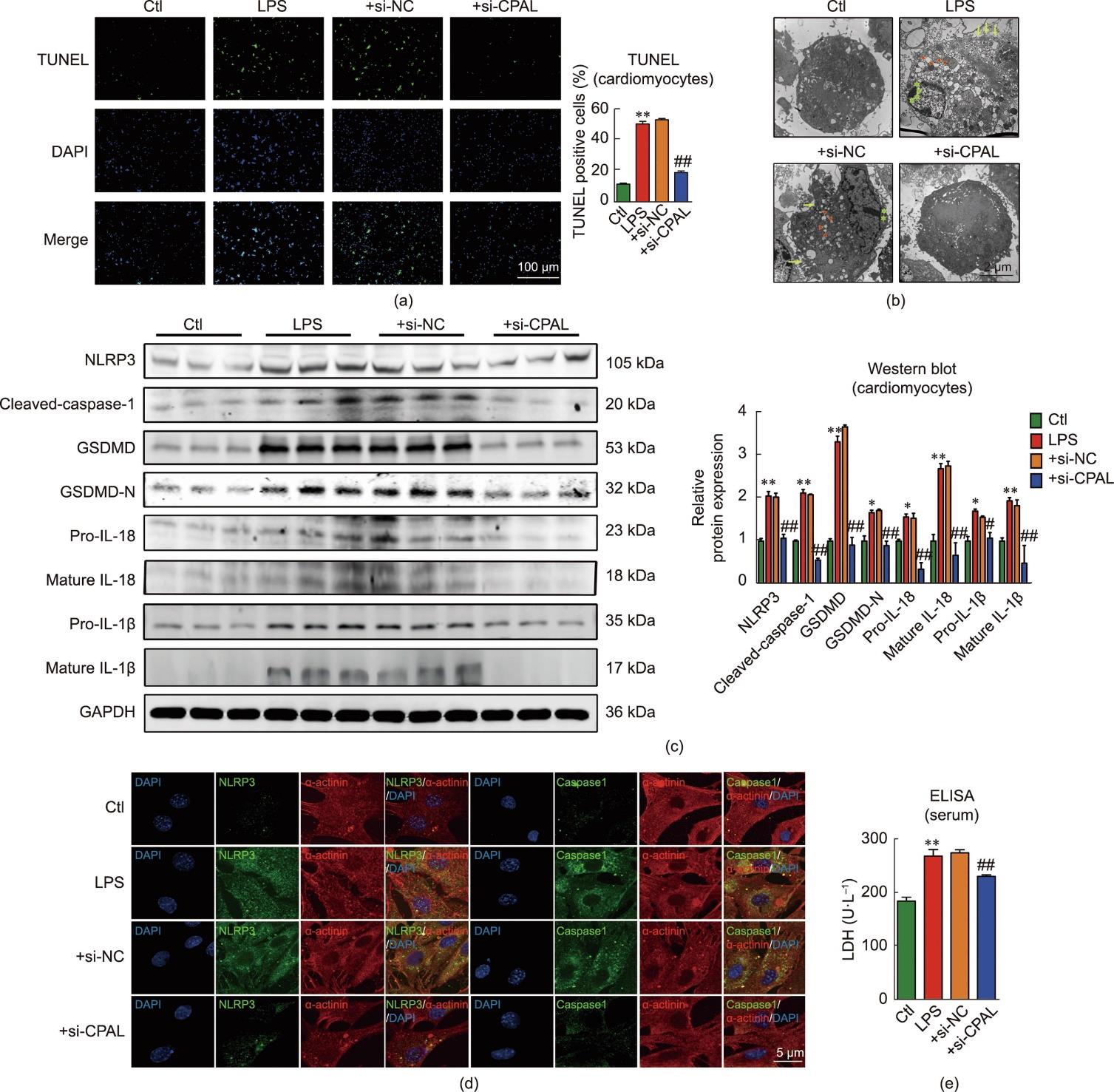
图6 沉默CPAL减少LPS诱导的心肌细胞焦亡。(a)TUNEL染色的代表性图像,对照组、LPS组、+si-NC组和+si-CPAL组中TUNEL阳性心肌细胞的百分比分析(绿色:TUNEL阳性细胞;蓝色:DAPI;标尺:100 μm;**P < 0.01,与对照组比较;##P < 0.01,与+si-NC组比较;n = 3~6;均值±标准误差)。(b)对照组、LPS组、+si-NC组和+si-CPAL组新生小鼠心肌细胞超微结构(绿色星号:核固缩;红色三角形:线粒体空泡;黄色箭头:膜溶解;标尺:2 μm;n = 3)。(c)对照组、LPS组、+si-NC组和+si-CPAL组中NLRP3、cleaved caspase-1、GSDMD、GSDMD-N、pro-IL-18、mature IL-18、pro-IL-1β 和mature IL-1β的蛋白质印记分析(n = 3~6;*P < 0.05,**P < 0.01,与对照组比较;#P < 0.05,##P < 0.01,与+si-NC组比较;均值±标准误差)。(d)对照组、LPS组、+si-NC组、+si-CPAL组新生小鼠心肌细胞中NLRP3/caspase-1-α辅肌动蛋白免疫荧光染色(绿色:NLRP3/caspase-1;红色:α辅肌动蛋白;蓝色:DAPI;标尺:5 μm;n = 4)。(e)各组细胞培养基中LDH的浓度检测(n = 4;**P < 0.01,与对照组比较;##P < 0.01,与+si-NC组比较;均值±标准误差)。
《3.7 CPAL是NFκB的上游激活因子》
3.7 CPAL是NFκB的上游激活因子
为了探究CPAL调节心肌细胞焦亡的关键机制,通过catRAPID数据库对RNA-蛋白结合可能性进行了生物信息学分析。结果显示,CPAL中两个特定区域,即序列76~127 nt和209~260 nt与NFκB相互作用的概率很高[图7(a)]。基于以上分析结果,进一步探讨CPAL与NFκB之间的调控关系。通过RIP实验来确定CPAL与NFκB之间是否存在直接结合关系,结果清楚地显示,NFκB免疫共沉淀物中含有多个CPAL特异性扩增产物,表明这种相互作用是存在的,且CPAL对NFκB有很强的特异性亲和力[图7(b)]。此外,探究了在这种直接结合的基础上,CPAL是否对NFκB活性具有调控作用。蛋白质印迹分析结果表明,敲减CPAL的小鼠梗死边缘区心肌组织中P-NFκB蛋白水平明显下调,而总NFκB蛋白表达无明显变化[图7(c)]。免疫组化结果表明,在CPAL敲减的心肌梗死小鼠梗死边缘区心肌组织中也观察到类似的P-NFκB下调的现象[图7(d)]。在体外,利用CPAL的siRNA来进一步验证CPAL对NFκB的调控作用。结果显示,与+si-NC组相比,+si-CPAL组P-NFκB蛋白水平表达明显下调[图7(e)],而总NFκB的蛋白表达没有变化。免疫荧光分析结果显示,LPS处理可促进P-NFκB p65在心肌细胞核内聚集,而沉默CPAL后可抑制这一现象[图7(f)])。以上结果提示,CPAL可能通过促进NFκB的磷酸化来激活NFκB功能,从而使NFκB进入细胞核。为了进一步明确CPAL调控NFκB磷酸化是否是通过site1(CTCGGAGAGACCCTGGAGGGACCAGGACCACTCTTGTCTCGCCTGGACCTGC)和site2(TGTCTGAAGAAAAATGCTGGCCACTAATCGTTTGCCTCGAAGGAGTCTGAGG)直接与NFκB结合而发挥功能,本文采用寡聚脱氧核苷酸(ODN)技术构建了两个特异性靶向来屏蔽NFκB结合位点的CPAL-ODN序列片段,分别标记为CPAL-ODN-1(屏蔽site1)和CPAL-ODN-2(屏蔽site2)。这两个ODN序列分别与两个预测的CPAL结合位点完全互补。结果显示,CPAL和ODN共转染后,未影响P-NFκB水平[图7(g)]。说明CPAL可与NFκB结合位点(site1: 76~127 nt和site2: 209~260 nt)直接结合并诱导NFκB磷酸化。
《图7》
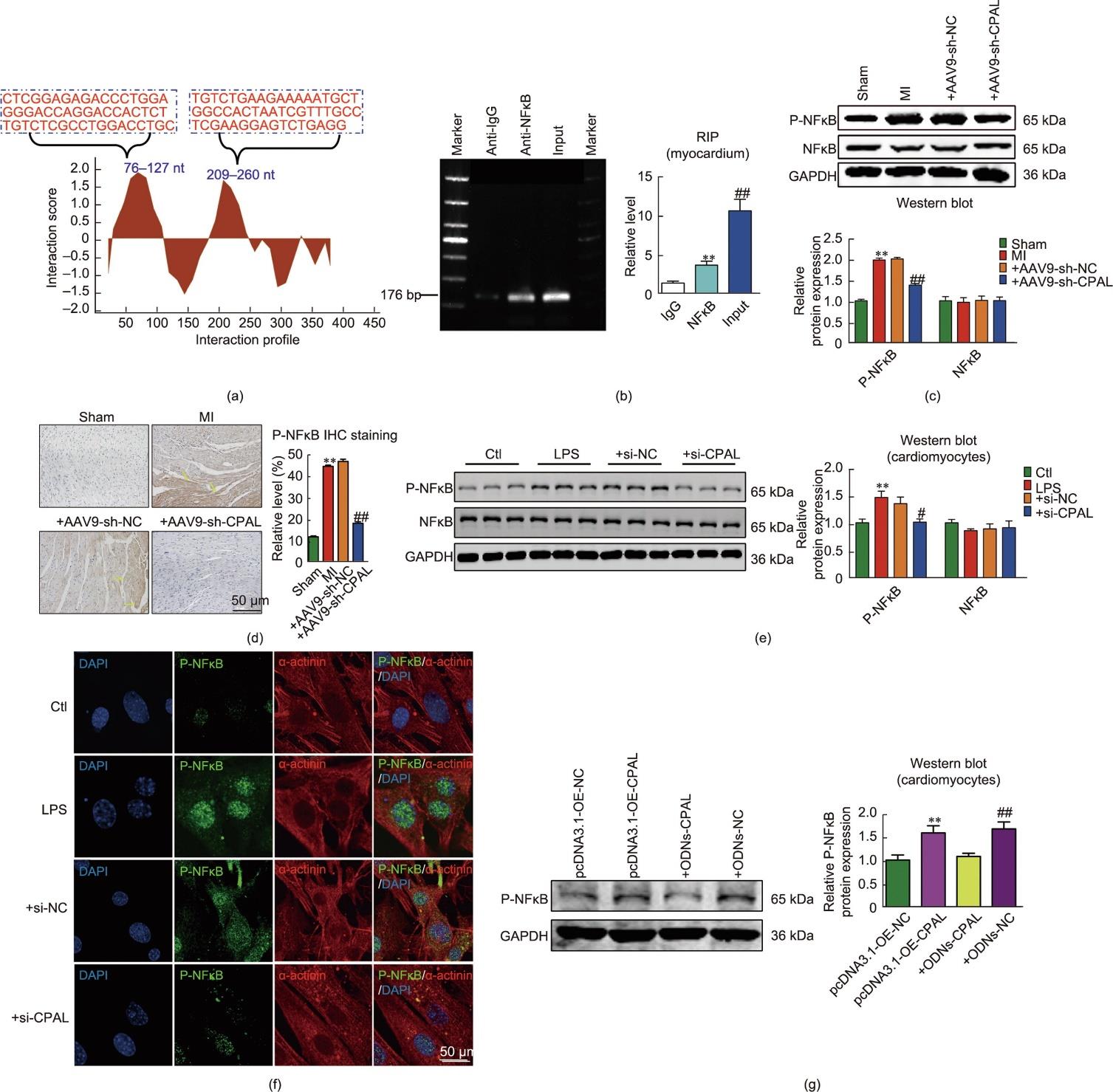
图7 CPAL是NFκB的上游调控因子。(a)通过在线预测网站发现CPAL与NFκB结合的两个特定区域(76~127 nt和209~260 nt)。(b)RIP实验分析CPAL与NFκB相互作用,NFκB免疫沉淀中检测到CPAL表达(**P < 0.01,与anti-IgG相比较;n = 3)。(c)P-NFκB、NFκB蛋白在各组中的表达水平(**P < 0.01,与sham组比较;##P < 0.01,与+AAV9-sh-NC组比较;n = 4~6)。(d)P-NFκB蛋白在左心室梗死边缘区免疫组化染色(标尺:50 µm;**P < 0.01,与sham组比较;##P < 0.01,与+AAV9-sh-NC组比较;n = 3~4)。(e)P-NFκB和NFκB蛋白在对照组、LPS组、+si-NC组、+si-CPAL组心肌细胞中的表达水平,GAPDH作为内参进行归一化(**P < 0.01,与对照组比较;#P < 0.05,与+si-NC组比较;n = 9。(f)对照组、LPS组、+si-NC组和+si-CPAL组心肌细胞P-NFκB(绿色)和α辅肌动蛋白(红色)免疫荧光染色(蓝色:DAPI;标尺:5 μm;n = 4)。(g)P-NFκB蛋白在pcDNA3.1-OE-NC组、pcDNA3.1-OE-CPAL组、+ODN-CPAL组、+ODN-NC组新生小鼠心肌细胞中的表达水平(**P < 0.01,与pcDNA3.1-OE-NC组比较;##P < 0.01,与+ODN-CPAL组比较;n = 5;均值±标准误差)。
《3.8 抑制NFκB可减少LPS诱导的小鼠心肌细胞焦亡》
3.8 抑制NFκB可减少LPS诱导的小鼠心肌细胞焦亡
为了探究NFκB在LPS诱导的小鼠原代心肌细胞焦亡中的作用,给予心肌细胞SN50(NFκB核易位的细胞渗透抑制剂)和Ac-YVAD-CMK(caspase-1抑制剂)处理[图8(b)]。蛋白质印迹实验结果表明,SN50在LPS处理的原代心肌细胞中显著抑制pro-caspase-1和cleaved caspase-1的蛋白表达水平,表明caspase-1很可能是NFκB转录调控的下游基因。同时,发现Ac-YVAD-CMK可明显抑制心肌细胞焦亡相关基因NLRP3、pro-caspase-1、cleaved caspase-1、GSDMD、GSDMD-N、pro-IL-18、mature IL-18、pro-IL-1β、mature IL-1β蛋白的表达水平,说明心肌细胞中caspase-1抑制剂显著抑制焦亡相关基因的表达。两种抑制剂共同作用时,心肌细胞中NLRP3、pro-caspase-1、cleaved caspase-1、GSDMD、GSDMD-N、pro-IL-18、mature IL-18、pro-IL-1β、mature IL-1β蛋白的表达水平明显降低,提示NFκB通过调控心肌细胞caspase-1的表达参与炎症反应和焦亡。最后,探究NFκB是如何调控caspase-1的表达,是否对caspase-1活性有直接调控作用。因此,利用TF-Protein Interactiotn Prediction(PROMO)数据库对DNA-蛋白质结合进行了理论分析。结果表明,Caspase-1 DNA启动子的近端区域(site1和site2)附近存在NFκB的结合序列,且这些序列在人、大鼠和小鼠中是保守的。ChIP实验结果显示,NFκB被募集到Caspase-1的近端启动子区域site1和site2,表明NFκB与Caspase-1有很强的亲和力[图8(b)]。为了进一步证实caspase-1的表达升高是由NFκB同时直接结合到site1和site2上所介导的,本研究采用了ODN技术,根据预测的两个caspase-1结合位点,分别设计了两个caspase1-ODN序列片段,即caspase1-ODN-1(屏蔽site1)和caspase1-ODN-2(屏蔽site2),分别靶向屏蔽NFκB的结合位点。在LPS处理的原代心肌细胞中共转染ODN-caspase1后,LPS处理未能调控pro-caspase-1、cleaved caspase-1、GSDMD-N的表达水平[图8(c)]。这说明NFκB能够与Caspase-1直接结合,并通过结合位点(site1: -846~836 bp和site2: -1544~1555 bp)调控Caspase-1转录。
《图8》

图8 抑制NFκB可减轻LPS诱导的心肌细胞焦亡。(a)LPS条件下SN50(5 µg∙mL-1, 24 h)和Ac-YVAD-CMK(50 µg∙mL-1, 24 h)处理后NLRP3、cleaved caspase-1、GSDMD、GSDMD-N、pro-IL-18、mature IL-18、pro-IL-1β和mature IL-1β蛋白表达水平(*P < 0.05,**P < 0.01,与对照组比较;#P < 0.05,##P < 0.01,与LPS组比较;n = 3~5;均值±标准误差)。(b)NFκB通过结合caspase-1启动子正向激活caspase-1。(左)caspase-1启动子中保守的NFκB DNA结合位点。PROMO工具显示caspase-1启动子近端区域上游2000 bp处的潜在结合位点。(右)ChIP实验结果,NFκB募集到caspase-1 site1和site2的近端启动子区域(n = 4)。(c)cleaved caspase-1、pro-caspase-1和GSDMD-N蛋白在对照组、LPS组、+ODN-caspase1组和+ODN-NC组新生小鼠心肌细胞中的表达水平(*P < 0.05,**P < 0.01,与对照组比较;#P < 0.01,##P < 0.01,与LPS组比较;n = 3~4)。
《4、 讨论》
4、 讨论
本研究首次发现lncRNA CPAL的关键病理生理学意义及其在心肌梗死中的精确调控机制。研究表明,CPAL在心肌梗死小鼠梗死边缘区心肌组织中上调,并导致了心肌重塑和心脏功能障碍。而敲减内源性CPAL可部分抑制缺血心肌的糖脂质代谢改变,并减少心肌梗死后心肌细胞焦亡的现象。此外,发现CPAL可直接与NFκB蛋白相互作用并增加其磷酸化水平,磷酸化的NFκB可以从细胞质转运到细胞核中,并增加下游基因转录活性;这可能是CPAL调控心肌梗死后心脏代谢改变和焦亡的基础。
在心肌梗死的过程中,受到缺血刺激的心肌细胞会出现能量代谢紊乱、氧化应激、细胞死亡及炎症等。葡萄糖和脂质代谢紊乱在糖尿病和其他疾病的心脏重塑发展中起着重要作用[31]。已有研究表明,心脏中葡萄糖和脂质代谢的变化会引发心肌细胞凋亡、心肌肥大和心脏纤维化[31]。此外,炎症也参与了梗死心脏的修复和重塑[32],炎症反应的连续活动模式会释放炎症细胞因子,引起代谢紊乱,从而加速心肌梗死的心肌细胞重塑[33]。在这项研究中,发现心肌梗死后梗死边缘区心肌组织中ATP生成减少,CD36、HK1和GLUT4的表达异常,并伴有心肌细胞炎症损伤。这些数据表明,心肌梗死后小鼠的心肌细胞中存在葡萄糖和脂质代谢紊乱以及心肌细胞焦亡的现象。
最近的研究表明,lncRNA是不同器官中代谢反应和生理平衡的重要调节因子[34]。然而,参与心肌梗死后心脏能量代谢和心肌炎症过程的lncRNA还没有被发现。在本研究中,发现CPAL是心肌梗死相关的新的lncRNA,在人、兔和小鼠中都是保守的,且在心肌梗死小鼠梗死边缘区心肌组织中表达增加。研究结果表明,CPAL在心肌梗死后心脏重塑过程中对葡萄糖和脂质代谢紊乱和炎症过程的调节具有关键作用。更具体地说,沉默CPAL可以减弱小鼠心肌梗死后心脏的糖脂代谢紊乱和心肌细胞焦亡,表明CPAL是预防心肌梗死后心脏损伤的新治疗靶标。
NFκB是血管内皮细胞和心肌细胞中重要的转录因子[35]。它与IκB(一种抑制蛋白)结合,IκB在正常生理条件下以同源二聚体的形式存在,不具有活性[36‒37]。在焦亡激活过程中,Nod样受体(NLR)、pyrin、AIM2与ASC结构域结合,激活pro-caspase-1,生成有活性的caspase-1 [38]。lncRNA通常通过共享相同的转录因子与mRNA相互作用[8]。有报道称lncRNA MALAT1作为ceRNA抑制NFκB的信号通路,其表达随着衰老的程度而逐渐降低[39]。在本研究中,发现lncRNA CPAL与NFκB结合,抑制NFκB与IκB复合物的形成,并通过使Ser536位点的磷酸化激活NFκB,促进P-NFκB从细胞质转移到细胞核,在细胞核中与特定的pro-caspase-1结合,来调节其转录活性。此外,已有研究表明,NLRP3是细胞内蛋白NLR的家族成员之一,在激活含有caspase-1的复合物(炎症小体)中起着重要作用[39‒40]。有报道显示NFκB的激活依赖于TLR诱导的NLRP3的表达增加[41]。本文研究结果证明,CPAL的缺失减弱了NFκB的磷酸化作用,抑制了NLRP3的表达。但这一点还需要进一步的研究来证实,后续将继续研究CPAL与NFκB磷酸化之间作用关系。
此外,NFκB是心肌糖脂代谢的关键调控因子。NFκB的下游靶基因也参与糖脂和能量代谢。Zhu等[16]报道了QNZ通过抑制NFκB活性,促进GLUT4蛋白表达改善葡萄糖摄取,抑制软骨细胞退变。Wu等[42]发现LPS通过NFκB/Snail/HK3信号通路促进结直肠癌细胞糖酵解和转移。Han等[43]认为,高水平的CD36通过NFκB介导的脂质积累和代谢重编程来促进炎症反应。在本研究中发现CPAL缺失通过调节缺血心肌中GLUT4、HK1和CD36蛋白的表达,增强了葡萄糖和脂肪酸的利用,改善了能量代谢。综上所述,本研究揭示了CPAL作为一种有害lncRNA,通过靶向P-NFκB p65信号通路,促进心肌梗死过程中糖脂代谢紊乱和心肌细胞焦亡。目前尚不清楚CPAL介导的代谢改变和NFκB介导的心肌细胞焦亡的之间的先后顺序,还有待进一步探讨和研究。
《5、 结论》
5、 结论
本研究结果表明,CPAL不仅可以作为心肌梗死的生物标志物或预测因子,而且可能是心肌梗死后心肌细胞焦亡、糖脂代谢异常以及其他心血管疾病的关键调节因子。本研究旨在评估CPAL缺乏对心脏功能的保护作用,为改善缺血/炎症引起的心脏功能障碍提供了一个令人兴奋的途径。揭示CPAL可能是改善心肌梗死患者心脏功能的关键治疗靶点。











 京公网安备 11010502051620号
京公网安备 11010502051620号




