

《1、 引言》
1、 引言
现代化工产业是我国的碳排放大户,是实现碳中和目标需要重点关注的行业。在过去的10年里,全球煤化工、石油化工等行业产生的二氧化碳(CO2)排放量增加了50多倍(从每年9.1亿吨增加到每年490亿吨),我国行业发展与环境保护之间的矛盾也日益加剧[1‒2]。近年,我国提出碳中和长期目标,即CO2排放量力争在2030年前达到峰值,并在2060年前实现碳中和。碳中和意味着未来我国的碳排放要和碳汇(指植物吸收CO2和人工捕集CO2总量)相等。据统计,我国目前的碳汇只有每年不到15亿吨,还明显低于化学工业的碳排放量[2]。现代化工要实现碳中和远期目标面临巨大挑战。
以煤化工行业为例,在现代煤化工生产中,原煤中的碳元素最终只有1/5~1/3进入产品;其余大部分都成为了巨量碳排放的源头[2]。此外,在煤化工生产过程中,半数的能源消耗以各种形式的余热被直接废弃,成为环境热污染。因此,在大幅降低煤、石油等传统化石能源在我国能源结构中占比的同时,通过CO2的资源化转化和化工余热的高效利用,将更多的碳元素和能源转化进入产品,对实现碳中和目标至关重要[3]。最近,学术界对基于固体氧化物电解池(SOEC)的高温电解技术给予了高度关注[1,4]。结合我国西北地区丰富的风电、光电等可再生电能,SOEC可以充分利用化工余热,将CO2通过电解反应转化为高附加值的燃料和化工产品,同步实现可再生能源的大规模储存以及CO2的资源化利用(图1)[4‒6]。更重要的是,与其他低温(≤200 ℃)转化的电化学技术(如基于液体电解质的电解技术)相比,在800~1000 ℃的高温下运行的SOEC具有许多独特的优势,包括总阻抗低、耗电量低、电解效率高以及对贵金属电极催化剂的依赖性更小[2]。此外,即使在较大的电流密度下,SOEC也可以稳定运行。例如,碱性电解槽(AEC)的稳定工作电流密度通常低于0.5 A∙cm-2,而最先进的SOEC的稳定工作电流往往高于1.0 A∙cm-2,且在工作条件下极化损耗低,性能衰减可忽略不计[4,7]。这意味着,为了达到相同电解能力,所需SOEC电解池堆的体积远小于AEC电解池堆。因此,SOEC电解池堆更便于实现模块化灵活设计,并应用于某些特定场景之中[2,7]。
《图1》

图1 基于高温电解的CO2高效转化和利用过程示意图[
《2、 用于CO转化的固体氧化物电解池概述》
2、 用于CO转化的固体氧化物电解池概述
SOEC在高温下转化CO2的方法主要有两大类:一类是纯CO2电解制CO和O2;另一类是在其他物质存在时的CO2电解,简称共电解,如CO2/H2O共电解和CO2/CH4共电解[1]。本文讨论的范围既包括纯CO2电解,也包括共电解。使用SOEC进行CO2转化的基本原理如图2所示(以纯CO2电解为例)。通常,一个电解池基本单元由阴极、电解质和阳极组成。在CO2电解(或共电解)过程中,CO2(或CO2/H2O)分子在阴极/电解质界面被还原,产生气态CO(或CO/H2)和氧离子O2-。这些在阴极产生的气体随载气通过多孔电极;产生的氧离子O2-则在高温下迁移通过致密的电解质层,随后在电解质/阳极界面放电,生成气态O2 [8‒12]。主要反应式包括:
《图2》

图2 用于CO2转化的固体氧化物电解池的基本原理示意图。
阴极:
(1)
阳极:
(2)
总反应:
(3)
CO2/H2O共电解不仅可以实现电能向化学能(CO/H2,即合成气)的有效转化,而且实现了CO2资源化利用,因此这项技术吸引了许多研究者的关注[2,12]。相较于纯水蒸气电解而言,CO2/H2O共电解要复杂得多,因为该体系中存在逆水煤气变换(RWGS)反应,即H2O在正向电解反应中被还原成H2,随后CO2再被H2还原[2,13]:
(4)
根据现有文献结果,RWGS反应的吉布斯自由能变化(
与共电解相比,纯CO2电解对水资源的依赖较少,因而适用范围更广,对于实现碳中和目标也具有更大的意义。然而,它也具有更高的技术门槛,现阶段面临的挑战更多[2,12]。未来,随着电化学基础理论的发展和相关技术的进步,CO2电解有望发展成为一项极富前景的新兴工业化技术[1‒2]。美国航空航天局(NASA)在其火星探索项目中,就曾提出采用SOEC电解火星大气中丰富的CO2以制造氧气推进剂,供太空探测车使用[15]。
尽管CO2电解技术或共电解技术具有广阔的应用前景,但由于CO2的分子结构远比H2O分子稳定,因而CO2电解反应无论是在热力学还是在动力学上都比H2O电解反应更加难以发生,这对SOEC组件材料的电催化活性和电解运行稳定性提出了更高的要求,目前的产业化进程也相对更为缓慢。为此,应当针对性地开发具有相对较高电化学活性和运行稳定性的新型SOEC组分材料,以实现稳定的CO2电解或共电解过程[16]。在本文接下来的章节中,我们分别介绍了适用于CO2电解或共电解的SOEC阴极、电解质、阳极等电解池组件材料的最新研究进展,并详细讨论了SOEC在发生CO2电解过程中的电化学界面反应基元反应步骤和关键机理。此外,本文结合CO2电解过程的特点,探讨了SOEC高温电解技术与化工合成过程相耦合以实现碳中和目标的方法。本文的讨论结果可为高性能SOEC元件的合理设计和优化以及工业规模SOEC电堆与模块的设计和应用提供参考。
《3、 SOEC的关键材料》
3、 SOEC的关键材料
《3.1 阴极材料》
3.1 阴极材料
在CO2电解过程中,CO2分子在SOEC阴极侧放电,发生碳还原反应并生成氧离子(CO2 + 2e-
《3.1.1. 金属复合阴极材料》
3.1.1. 金属复合阴极材料
以镍-氧化钇稳定氧化锆(Ni-YSZ)为代表的一系列金属-陶瓷氧化物复合材料,是目前研究最为广泛的一类SOEC阴极材料。在制备这类电极时,最为经典的方法是将氧化镍(NiO)、YSZ与造孔剂[如淀粉、聚甲基丙烯酸甲酯(PMMA)等]共烧,之后在H2气氛下还原,得到具有多孔形貌的Ni-YSZ复合电极[2]。在Ni-YSZ复合电极中,金属Ni提供电子导电性,并可为CO2的化学吸附和电化学还原提供反应位点;YSZ陶瓷可提供离子导电性,且它在高温烧结后具有较高的机械强度,可以起到支撑阴极的作用;造孔剂在高温烧结后会在电极体相留下大量孔隙,可为H2O、CO2、CO等气体小分子的扩散提供通道。
目前,Ni-YSZ材料由于具有较好的电化学性能和稳定性,发展已经较为成熟,在CO2电解和CO2/H2O共电解领域也已经有了广泛的应用。早在2007年,丹麦Risø国家实验室就已报道其开发的Ni-YSZ|YSZ|LSM-YSZ电解池堆在950 ℃、70% CO2 + 30% CO混合气氛中可实现稳定的CO2电解,运行电流密度为1.5 A·cm-2,电解电压为1.29 V,CO产量可达33 L∙h-1 [9]。在另一项丹麦Risø国家实验室与Topsøe公司共同开展的研究中,研究者将10片装12 cm × 12 cm的Ni-YSZ|YSZ|LSM-YSZ电解池堆置于850 ℃、45% H2O + 45% CO2 + 10% H2混合气氛中,在0.50 A·cm-2和0.75 A·cm-2电流密度下分别稳定电解运行了800 h和400 h,均未发现电解池性能衰减迹象[17]。尽管如此,也有研究表明,Ni-YSZ阴极在更大电流密度或在更长时间持续运行过程中可能会出现衰减。德国Jülich研究所考察了Ni-YSZ|YSZ|LSCF电解池堆在H2O/CO2/H2混合气氛下的长期共电解性能,发现其在0.300~0.875 A∙cm-2电流密度区间内的性能衰减速率约为2%~4%/1000 h;当电解电流密度提升至0.875 A∙cm-2以上时,性能衰减速率更会高达约6%/1000 h [18]。
许多研究者对Ni-YSZ阴极在CO2电解运行过程中的衰减机理进行了研究。Hauch等[19]利用阻抗谱法对Ni-YSZ|YSZ|LSM-YSZ电解池堆进行分析表征,证明了电解池性能的衰减主要是由Ni-YSZ阴极的极化阻抗增加所引起的;特别地,他们发现在高温(950 ℃)、大电流密度(2 A·cm-2)运行条件下,金属Ni颗粒会发生迁移和团聚,显著地改变阴极-电解质界面结构(如形成Ni致密层或YSZ致密层),从而极大地增加阴极阻抗。Skafte等[5]通过原位X射线光电子能谱(XPS)表征了Ni-YSZ薄膜电极在CO/CO2气氛中的衰减情况,发现在750 ℃、气氛中CO分压约为73%的条件下,当电极过电位提升至150 mV时,电极表面就会出现明显的C‒C sp2及C‒C sp3信号,此时通过扫描电镜可发现清晰的表面积碳现象[图3(a)] [5]。Argyle和Bartholomew [20]指出,沉积的碳单质可能来源于CO在金属Ni表面发生的歧化反应。
《图3》

图3 (a)碳沉积后Ni-YSZ表面的扫描电镜图像;(b)当阴极开始在CO/CO2气氛中沉积碳时,三种电极(Ni-YSZ、Ni-SDC和SDC)在过电位处的XPS峰[
为了提升Ni-YSZ阴极在高温、大电流密度下电解CO2的电催化活性和运行稳定性,许多研究者对其进行了优化改性,主要手段包括无机金属氧化物修饰等[5,21‒24]。在Skafte等[5]的原位XPS研究中发现,若采用Sm0.2Ce0.8O1.9-
许多研究者对SOEC系统中的CO2高温电化学还原反应过程和机理展开了深入的研究,研究结果揭示了SOEC在电解过程中产生积碳的原因,也为Ni-YSZ基阴极材料的设计优化提供了指导[5,20]。如图4(a)所示,目前一般认为,在Ni-YSZ表面上CO2高温电化学还原反应包括以下基元步骤:①气态CO2分子在金属Ni表面的吸附和活化(
《图4》
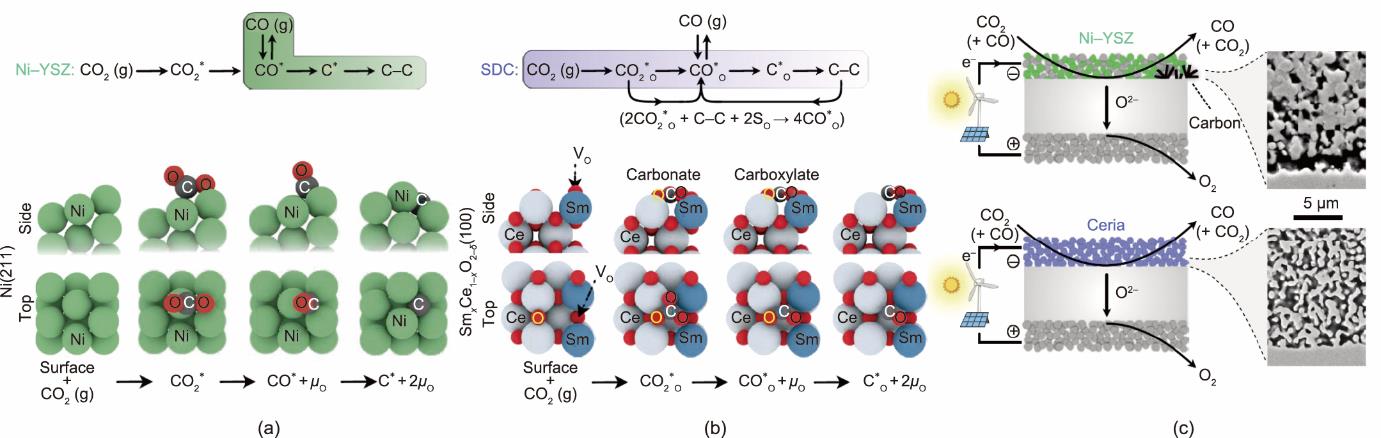
图4 (a)、(b)SOEC运行条件下Ni-YSZ和SDC表面CO2还原机理示意图;(c)两种类型的阴极/电解质界面的横截面扫描电镜图像[
CeO2及其衍生物是一种可作为SOEC阴极的备选材料,它可以显著增加阴极在含碳气氛中长期工作的稳定性[21‒24]。这是由于铈基电极材料(如Gd
(5)
对比Ni-YSZ和SDC表面上的积碳过程和反应机理,可以充分看出,SDC基电极材料在高温电解条件下具有显著更高的抗积碳特性。这种极高的抗积碳特性也可以通过观测两种类型的阴极/电解质界面的横截面扫描电镜图像来进一步证实,如图4(c)所示。上述实验和讨论结果都表明,二氧化铈及其衍生物是具有潜力的电极催化材料,有望进一步被开发成为适用于CO2电化学还原的SOEC阴极。
Neagu等[23]和Yue等[24]研究发现,Ni-YSZ表面的积碳反应速率还强烈依赖于复合电极中金属Ni晶粒的尺寸大小。Christensen等[25]和Chen等[26]认为,在较小的Ni晶粒中,碳的饱和浓度相对更高,因此电极对于碳单质的容纳程度更高。按照此思路,通过开发电极制备新工艺,降低Ni-YSZ电极中金属Ni的平均粒径,可能是一条提升电极抗积碳性能的有效策略。Han等[27]通过[Ni(acac)2]前驱体还原制备了均匀分布在二氧化硅表面的Ni纳米颗粒,它们的平均粒径仅为(5.2±0.4) nm;这些Ni纳米颗粒在800 ℃、CH4/CO2气氛中处理170 h后,表面未出现任何积碳现象。Liang等[28]开发了尿素燃烧原位合成工艺来制备NiO-YSZ电极材料前驱体(其中,NiO的粒径约为11 nm),经H2还原后,可以生成细小且在YSZ基体中均匀分散的Ni金属颗粒。然而,通过这些方法制备的金属纳米颗粒的热稳定性和化学稳定性仍需要进一步提高,且在这样的电极材料体系中,金属纳米粒子的团聚可能成为引起电极性能衰减的主要因素。
还有一类可被用作SOEC阴极的重要材料是含铜复合材料[29‒32],典型代表就是Cu-CeO2-YSZ及其衍生物。这类材料已被广泛用于催化CO2高温电解反应,因为其表面不易发生积碳反应,并且,将这类材料组成的电极暴露于CO2或CO气氛时,通常还会表现出电池开路电压(OCV)小幅降低的现象[32‒33]。Cheng等[34]通过在YSZ衬底上烧结Cu/Gd0.1Ce0.9O2 (CGO)浆料的方法,在YSZ电解质上制备了多孔Cu-CGO电极黏附层,并以此作为阴极,制备了Cu-CGO|YSZ|YSZ-LSM|LSM电解池;该电解池在750 ℃、1:1 CO2/CO气氛条件下还原CO2的2 h期间,没有出现性能下降情况。在另一项研究中,Su等[29]以YSZ为电解质载体,采用浸渍法在其表面制备了Cu-SDC电极,形成了Cu-SDC|YSZ|Cu-SDC对称电池。在CO/CO2(50:50)以及CO/CO2(67:33)气氛下,用该电池进行CO2电解还原的活化能仅约为1.57 eV;向电极中浸渍更多的Cu元素,可产生更多的Cu/CeO
《3.1.2. 钙钛矿基阴极材料》
3.1.2. 钙钛矿基阴极材料
近年来,人们发现许多钙钛矿基氧化物具有混合电子-离子传导性能(MIEC)和较好的电催化活性,且在氧化-还原循环稳定性及抗积碳性能上优于传统的Ni-YSZ金属陶瓷复合电极材料,有望成为SOEC阴极材料的新选择[35‒37]。几类典型的钙钛矿基氧化物包括钙钛矿型材料(通式为ABO3-
《图5》

图5 几种钙钛矿基氧化物的晶体结构示意图。(a)钙钛矿材料;(b)Ruddlesden-Popper材料;(c)双钙钛矿材料[
LSCM在CO/CO2气氛中具有可观的离子电导率、电子电导率和优异的耐久性,是用于CO2电解或共电解的最常见的钙钛矿基SOEC阴极材料之一[39‒40,43]。为了进一步增强LSCM阴极的离子导电性,还经常会向其中添加YSZ或GDC [40]。Yue和Irvine [44]利用真空浸渗法,制备了Pd-GDC共负载的LSCM阴极,在900 ℃、CO/CO2(30:70)气氛下,该电极的极化电阻仅为0.24 Ω∙cm2。Zhang等[45]通过在多孔YSZ支架上共烧结LSCM和GDC纳米颗粒,制备了LSCM-GDC/YSZ复合阴极;在800 ℃和0.29 A∙cm-2的电流密度下,在阴极的50 h测试期间没有观察到颗粒聚集现象和性能衰减。最近Ma等[46]使用甘氨酸-硝酸盐法(GNP)合成了LSCM阴极,基于该阴极的LSCM|YSZ|LSCF全电池在800 ℃、1.5 V电解电压、H2O/CO2(60:40)气氛中可达到0.1 A∙cm-2的稳定运行电流密度,体现出其对CO2电解和共电解具有较好的稳定性。
LSF是适用于CO2高温电解的另一种重要的阴极材料[39‒40,47]。Yang等[48]开发了一个LSF|LSGM|LSCF单电池在纯CO2气氛中进行电解测试,在1.5 V、800 ℃时,电流密度达到了0.76 A∙cm-2。Zhou等[49]采用球磨和烧结等方法制备了Pd单原子嵌入的Pd-LSF-SDC阴极;基于该阴极的Pd-LSF-SDC|YSZ|LSM-YSZ单电池在1.6 V和800 ℃下电解纯CO2时,最高平均电流密度可达到0.58 A∙cm-2。上述结果表明,LSF及其衍生物是一种很有前景的高温CO2电解阴极材料。
此外,还有许多其他钙钛矿基材料在用于CO2高温电解时,也具有良好的性能,如LST、(La,Sr)VO3 (LSV)、Sr2Fe1.5Mo0.5O6-
《3.1.3. 表面纳米颗粒溶出的阴极材料》
3.1.3. 表面纳米颗粒溶出的阴极材料
近年来,有研究表明,在适当的还原性气氛条件下,钙钛矿基材料中的B位过渡金属离子可被充分还原,并以金属纳米颗粒或简单氧化物的形式在钙钛矿晶格表面析出,称为B位金属原位脱溶[图6(a)][41,50]。这些原位脱溶产生的金属纳米颗粒不仅具有优异的电子导电性和电化学催化活性,而且在高温、强还原性气氛条件下还具有特殊的稳定性,可以极大地提升钙钛矿阴极材料的电化学活性[23,51]。例如,Lv等[52]制备了双钙钛矿基Sr2Fe1.35Mo0.45Co0.2O6-
《图6》

图6 (a)钙钛矿阴极材料中B位阳离子脱溶示意图[
《图7》
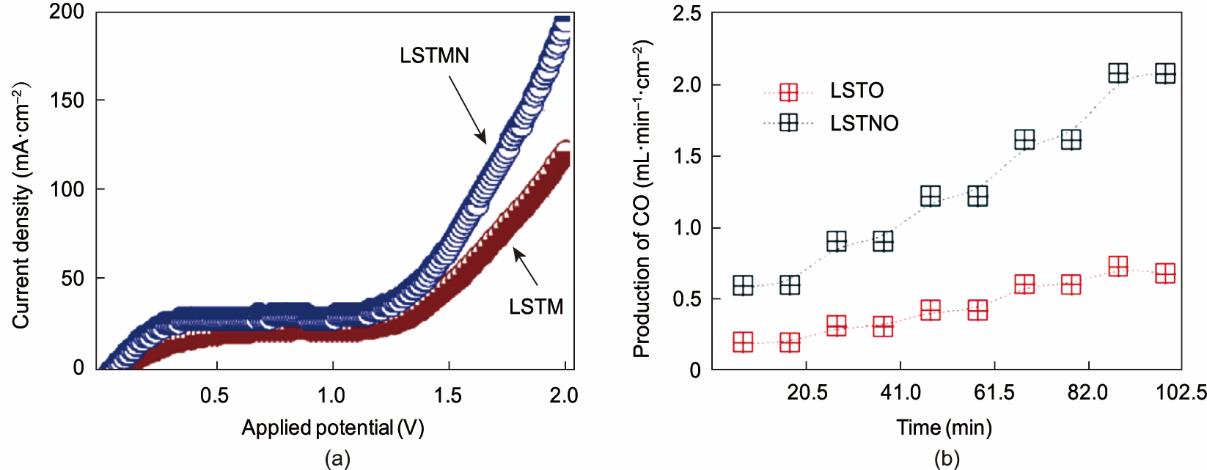
图7 (a)(La0.2Sr0.8)0.9(Ti0.9Mn0.1)0.9Ni0.1O3-δ阴极在Ni溶出前后的电化学性能对比[
许多研究表明,钙钛矿阴极表面由脱溶产生的纳米金属粒子不仅具有很高的电子导电性与电催化活性,而且在CO2电解运行条件下也具有优异的抗积碳性能[38,41]。Liu等[42]通过拉曼光谱对Co-Fe溶出的(Pr0.4Sr0.6)3(Fe0.85Mo0.15)2O7电极进行研究,结果表明,该阴极在850 ℃、70%CO2‒30%CO气氛下电解运行10 h后,电极表面未检测到石墨生成,而在同样条件下电解运行的Ni-YSZ阴极表面则出现了明显的石墨信号[图8(a)],这表明脱溶的Co-Fe纳米金属颗粒相较于传统的SOEC阴极具有出色的抗积碳特性。除此之外,Ni、Mn、Cr、Cu等脱溶金属纳米粒子的抗积碳稳定性也在其他SOEC阴极体系中得到证实[58‒61]。一些典型的脱溶阴极体系及其在长时间电解CO2运行过程中的主要性能指标汇总如表1 [42,52,55,58‒65]所示。Neagu等[23]认为,这些原位脱溶的金属纳米颗粒通常具有“锚定”在其原始位置的作用,其结构稳定性与化学稳定性远高于由浸渗等方法制备的复合材料体系。Zhao等[66]指出,锚定于阴极表面的金属纳米颗粒不仅可以促进CO2的电化学还原过程,而且可以通过快速电子交换和加速材料表面碳单质向含碳气相产物的转变来抑制电极表面积碳过程[图8(b)]。这一发现解释了这些阴极在CO2电解过程中具有特殊稳定性的原因。
《图8》
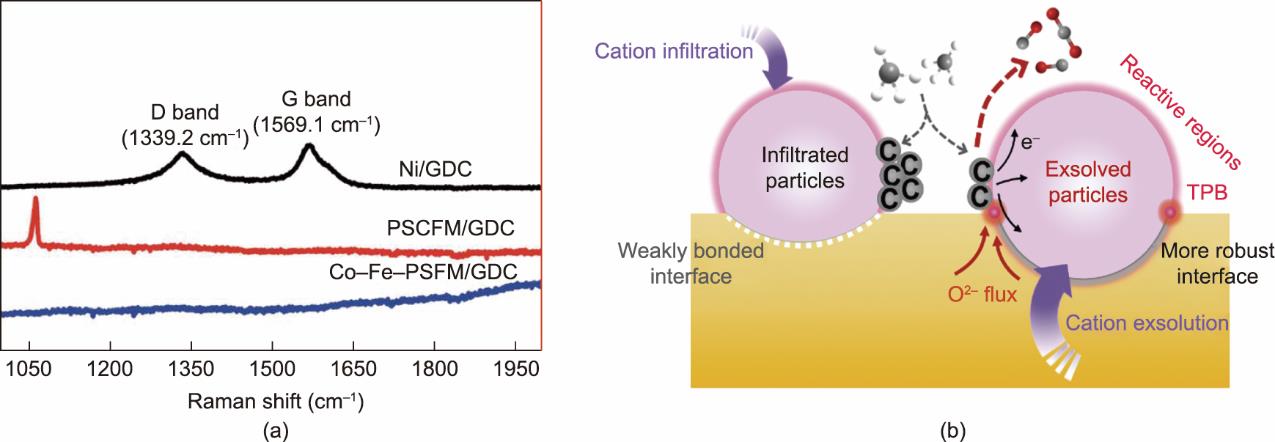
图8 (a)电解运行测试后的Ni/GDC、Pr0.4Sr0.6Co0.2Fe0.7Mo0.1O3‒δ (PSCFM)/GDC和Co-Fe-(Pr0.4Sr0.6)3(Fe0.85Mo0.15)2O7) (PSFM)/GDC阴极拉曼光谱[
《表1》
表1 典型的金属纳米粒子脱溶阴极在CO电解中的性能
| Exsolved component | Composition of perovskite matrix | Performance during CO2 electrolysis | Ref. |
|---|---|---|---|
| Fe | Sr2Fe1.5+ |
No carbon deposition after 100 h electrolysis at 850 ℃ under a CO2/CH4 atmosphere, 1.4 V electrolysis potential, and ~0.5 A∙cm-2 current density | [ |
| Co | Pr0.5Ba0.5Mn1‒ |
Stably electrolyzing under a CO/CO2 atmosphere; current density reaches 0.85, 1.6, and 2.5 A∙cm-2 at 800, 850, and 900 ℃, respectively | [ |
| Ni | La0.43Ca0.37Ni0.06Ti0.94O3 | No carbon deposition during 48 h electrolysis at 850 ℃ under a H2O/CO2 atmosphere at a current density of ~0.6 A∙cm-2; electrolysis potential only increased from 1.37 to 1.39 V during this process | [ |
| (La0.3Sr0.7)0.9Ti0.95Ni0.05O3‒ |
Stable CO2 electrolysis at 800 ℃ under a current density of ~0.3 A∙cm-2 and a 2 V potential in an H2O/CO2 atmosphere with 90% Faraday efficiency | [ |
|
| (La0.2Sr0.8)0.95Ti0.85Mn0.1Ni0.05O3+ |
Current density reaches 0.91 A∙cm-2 when electrolyzing pure CO2 at 850 ℃ and 2 V; no performance degradation during 10 redox cycles | [ |
|
| Mn | Nb1.33(Ti0.8Mn0.2)0.67O4 | Current density reaches ~1.6 A∙cm-2 when electrolyzing at 800 ℃, 1.6 V under a CO2/CO/Ar atmosphere; stably operating for more than 100 h at 1.4 V with no degradation | [ |
| Cr | Nb1.33(Ti0.8Cr0.2)0.67O4 | Current density reaches ~1.6 A∙cm-2 when electrolyzing at 800 ℃, 1.6 V under a CO2/CO/Ar atmosphere; stably operating for more than 100 h at 1.4 V with no degradation | [ |
| Ni-Cu alloy | NbTi0.5(Ni |
CO output reaches 0.1629 mL∙min-1∙cm-2 when running at 800 ℃, 1.4 V potential in a pure CO2 atmosphere | [ |
| Co-Fe alloy | (Pr0.4Sr0.6)3(Fe0.85Mo0.15)2O7 | No carbon deposits detected on the electrode surface after 10 h continuous electrolysis operation at 850 ℃ under a CO2-CO (70:30) atmosphere | [ |
| Sr2Fe1.35Mo0.45Co0.2O6- |
Stable CO2 electrolysis at 800 ℃ with a current density of 1.2 A∙cm-2 at 1.6 V, no performance degradation occurred in 12 oxidation-reduction cycles | [ |
|
| Fe-Ni alloy | La0.6Sr0.4Fe0.8Ni0.2O3- |
Current density reaches 1.78 A∙cm-2 when electrolyzing CO2/CO at 850 ℃ and 1.6 V; stably operating for more than 100 h at a current density of ~1.37 A∙cm-2 | [ |
| Fe-MnO | (Pr,Ba)Mn1- |
Stable electrolysis of pure CO2 at 850 ℃ with a current density of 638 mA∙cm-2 at 1.6 V | [ |
尽管关于金属纳米粒子脱溶增强钙钛矿电极电化学性能的研究已在SOEC阴极体系中得到了越来越广泛的应用,但金属纳米颗粒的脱溶机理以及驱动力尚不明晰。目前一般认为,钙钛矿中B位金属脱溶可能受钙钛矿晶格体相、表面缺陷分布情况及外部氧化还原性条件调控[38,41,67]。当钙钛矿晶格被还原时,其表面区域往往比体相更易形成氧空位,这些缺陷位点可通过电荷效应对晶体内部的B位过渡金属阳离子产生吸引作用;与此同时,表面缺陷的存在也降低了金属粒子成核的势垒,加速了B位过渡金属的还原和析出成核过程[23,41]。这些作用可持续驱动晶格内部的B位阳离子向表面迁移,并使表面脱溶纳米颗粒不断生长,直到达到平衡为止[23,67]。在未来,随着电极原位表征技术以及计算模拟手段的进一步发展,人们有望对钙钛矿中过渡金属离子的偏析机制和动力学过程形成更为明确的认识,从而做到对钙钛矿阴极脱溶组分和比例进行精准控制,用以指导设计开发具有高活性、高稳定性的SOEC阴极材料。
《3.2 阳极材料》
3.2 阳极材料
无论是对于水蒸气电解过程还是CO2电解过程,氧电极发生的氧析出反应(2O2-
目前,最广为使用的SOEC阳极材料包括以La1-
有研究表明,用化学性质相对稳定且具备一定配位性能的贵金属及其化合物对现有钙钛矿阳极材料进行修饰,可以同步提升阳极的电催化活性与运行稳定性。例如,Li等制备了La0.5Ba0.25Sr0.25Co0.8Fe0.2O3-
除了从微观层面上提升电极材料的电催化活性以及化学稳定性外,如何提升电极在宏观层面上的结构稳定性,同样也是人们研究和关注的重点。SOEC阳极发生的氧析出反应是一个气体分子数增加的过程,而目前人们常用的海绵状阳极中包含大量曲折孔或闭孔,气体扩散性能不佳,不利于氧气排出过程。尤其是当电解电流密度较大、产氧量较高时,在阳极内部容易形成局部高氧压位点,进而可能造成阳极/电解质界面脱层,导致电解池的极化阻抗急剧上升,危害电解池的长期运行稳定性[70,78]。因此,在改进SOEC氧电极材料活性的同时,降低阳极的气体传输与扩散阻力,提高电极结构稳定性和对大电流的抗衰减能力也是非常必要的。
为降低SOEC阳极气体传输与扩散阻力,许多研究者对阳极骨架结构进行了优化设计,并取得了显著的成效。例如,Chen等[79]通过冷冻流延法制备了具有微通道结构的Gd0.1Ce0.9O2-
《图9》

图9 (a)微通道电极SEM图像;(b)阳极体相氧气释放途径;(c)电解池电解极化曲线[
《3.3 电解质材料》
3.3 电解质材料
SOEC的电解质位于阴极和阳极之间,尽管不直接参与化学反应,但仍对电解池的整体性能起着至关重要的作用[82‒83]。电解质的作用主要是传导氧离子,同时阻隔电子传导和气体渗漏。为此,SOEC电解质材料一般应具有以下特性:①在运行条件下具有较高的离子电导率,但对于电子几乎不导通,以防止出现电解池内部短路现象;②与氢电极和氧电极材料的热膨胀系数基本匹配;③与氢电极和氧电极之间均不发生副反应;④在强氧化性/强还原性条件下都能保持结构稳定性和化学稳定性;⑤具有致密的形貌,以防止阴阳极两侧的气体相互扩散和渗漏[2]。
通常用于氧离子传导SOEC(称为O-SOEC)的电解质材料主要包括掺杂的锆基氧化物(如YSZ)以及铈基氧化物(如氧化钆掺杂氧化铈,GDC)和一些钙钛矿结构的氧化物(如La0.9Sr0.1Ga0.95Mg0.05O3-
《图10》
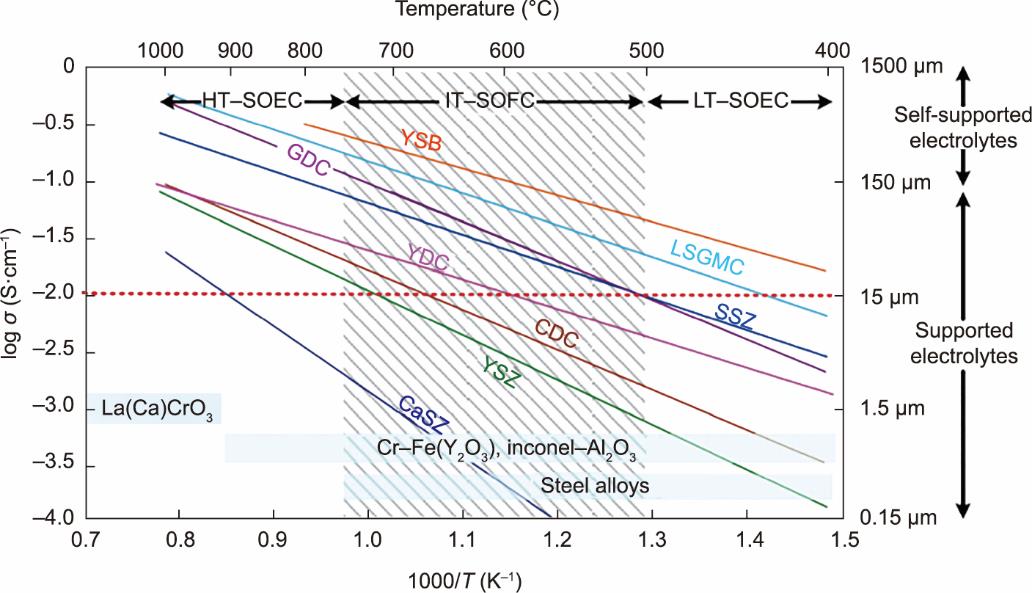
图10 典型的O-SOEC电解质材料及其导电性。HT:高温;IT:中温;LT:低温;YSB: (Bi2O3)0.75(Y2O3)0.25; LSGMC: LaxSr1-xGayMg1-y-zCozO3; SSZ: (ZrO2)0.8(Sc2O3)0.2; YDC: Ce0.8Y0.2O1.96; CDC: Ce0.9Ca0.1O1.8; CaSZ: Zr0.85Ca0.15O1.85 [
YSZ是用于高温SOEC的最为成熟的电解质材料之一,它具有优异的离子导电性、良好的稳定性和可接受的价格[84‒85]。然而,仍存在一些问题阻碍了YSZ材料进一步的工业化应用[2,62]。例如,许多研究人员指出,在致密的YSZ电解质层中,通常存在着许多晶界,晶界上的离子电导率比YSZ晶粒内部低1~2个数量级[84,86]。此外,当在高温下运行时,YSZ存在化学稳定性问题,可能与许多SOEC阳极成分(如LSM或LSC)发生反应,形成La2Zr2O7或SrZrO3等绝缘的第二相,从而导致电解质导电性的衰减[2,82,87]。为了解决这些问题,通常会向YSZ材料中掺杂铈基氧化物来提高其性能。Shen等[88]向YSZ电解质中掺杂了TiO2和SDC,发现在1000 ℃下,YSZ的离子电导率从0.09 S∙cm-1显著增加到0.123 S∙cm-1,这可能是因为这些掺杂组分的存在降低了YSZ的烧结温度,增加了晶粒尺寸。在另一项研究中,Kim等[89]制备了厚度为7 μm的Ce0.43Zr0.43Gd0.1Y0.04O2-
除了开发利用传统的YSZ之外,进一步探索新型高性能电解质材料,对于提高SOEC的整体性能也具有重要意义。最近,人们已经发现了一系列具有可观的离子导电性和在高温下优异的稳定性的钙钛矿基氧化物,它们有望被开发成为SOEC电解质材料。例如,La0.9Sr0.1Ga0.95Mg0.05O3‒
优化SOEC电解质的另一个研究方向就是减少其厚度,开发超薄而稳定的电解质层,以显著降低电解池的欧姆损耗,大大提高电解性能[11]。未来,分子束外延(MBE)、原子层沉积(ALD)、脉冲激光沉积(PLD)[85,93]等新制备技术的开发和应用,都可能为该领域带来革命性的转变,极大地加速CO2高温电解技术实现大规模产业化的步伐。
《4、 SOEC耦合化学减碳技术》
4、 SOEC耦合化学减碳技术
由前文讨论可知,SOEC器件所涉及的原材料大都具有价格便宜、在高温和含碳气氛下可保持结构和性能稳定等特点。这些特点不仅有利于SOEC技术在未来的大规模推广,而且也使得该器件可以适用于相对复杂的化工合成体系。通过将SOEC高温电解过程与化工合成过程进行耦合集成,得到各种燃料或化工原料,有望更大程度地发挥SOEC的价值。
《4.1 SOEC模块化设计与放大》
4.1 SOEC模块化设计与放大
为了满足不同规模的工业应用需求,需要对SOEC系统进行模块化设计。通过模块化设计,具有均匀尺寸的单个电池可以排列在一起形成SOEC电堆,数个电堆可以进一步组装成为SOEC模块(图11)[94]。一般来说,模块数量的增加,可以使电解池的总表面积增加,从而提高生产能力。因此,模块化设计可以扩大SOEC的尺寸,促进其工业应用,还可同时增加SOEC系统的灵活性和可靠性。
《图11》
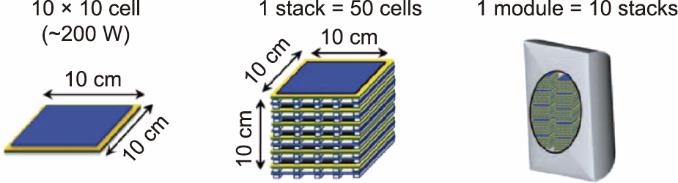
图11 将大小一致的单个电解池单元排列形成电堆和模块示意图[
应该注意的是,在复杂环境中运行的SOEC系统除了电堆本身之外,还应该包括许多必要的组件。例如,热管理系统,用于回收出口产品中包含的热量,从而提高整体电解效率;净化系统,用于调节入口气体的成分,并去除可能使烟囱衰减的杂质;自动控制系统,用于远程控制SOEC模块等[2,4]。近年来,这些系统配件相关的技术也经历了巨大的发展,然而,它们仍然存在着不稳定性和不可靠性,甚至这些系统组件的不可靠性才是SOEC系统出现故障的主要原因[4]。因此,在未来,为了进一步提高SOEC电堆和模块的运行稳定性和可靠性,开发和优化这些系统附件仍然是必要的。
《4.2 SOEC在化工行业的应用场景》
4.2 SOEC在化工行业的应用场景
在SOEC与化工耦合应用场景中,最广为人知的技术路径之一就是通过SOEC电解化工过程中产生的高温CO2/H2O废气,得到CO/H2合成气,再耦合费托合成反应,直接将合成气转化成为液态烃、醇、醛、酸等化工原料[1‒2,14]。该路径不仅可以有效利用化工过程产生的废气和余热,而且可将气氛中的CO2转化为液态含碳产物进行固碳,对于我国实现碳达峰、碳中和目标具有重要意义。在实验验证方面,Chen等[14]设计了一种管式SOEC-费托合成装置,将管式SOEC高温电解单元(LSM-YSZ|YSZ|Ni-YSZ管式电解池,操作温度约为800 ℃)与费托合成单元(操作温度约为250 ℃)相耦合,如图12所示。在进气量约为15 mL∙min-1 CO2/H2(CO2:H2=1:6,相对湿度为20%)的条件下,出口处甲烷的产率为0.84 mL∙min-1,CO2转换率达到64%。Luo等[95]制备了一种LSGM电解质支撑的管状SOEC,可实现一步法生产CH4,且从CO2到CH4的转化率高达98.7%。最近,Lee等[96]开发了一个六电池平管结构Ni-YSZ|YSZ-GDC|LSCF-GDC电堆,总活性面积为240 cm2,在800 ℃、进气H2O:CO2=2:1的条件下实现了连续500 h的稳定共电解运行。这些结果初步证明了该技术路径在实验室规模上的可行性。
《图12》
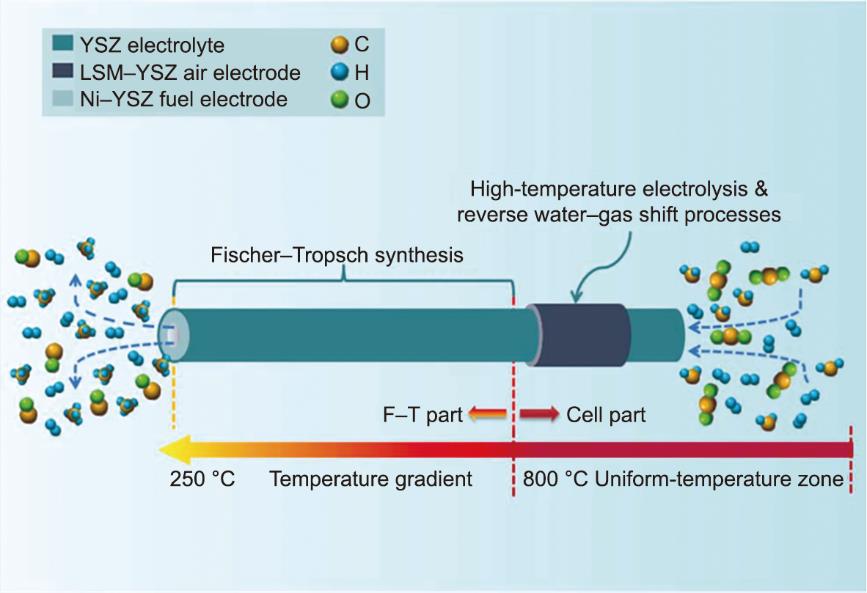
图12 SOEC耦合费托反应器实现CO2-H2O共电解直接合成甲烷示意图[
近年来,随着相关技术的不断发展优化,基于SOEC的CO2高温电解技术已经逐步走向工业化,并且其规模化进展迅速。2017年,来自Haldor Topsøe A/S公司(丹麦)的Küngas等[97]报道了世界上第一个商用CO2电解系统(eCOs),该系统可以电解产生99.995%纯度的CO,产量达到10 Nm3∙h-1以上。两年后,他们进一步对系统组件进行了改进,并提供了优化后的电堆测试数据:在750 ℃和-70 A的条件下,单个SOEC电堆的CO生产能力可达到2.2 Nm3∙h-1,且在超过5000 h运行期间,没有检测到性能衰减迹象[98]。基于这些进展,Haldor Topsøe A/S公司准备在未来两年内开发一系列商业化电解装置,这些装置的CO生产能力可达到96 Nm3∙h-1,纯度超过99.95% [97‒98]。2019年,Sunfire GmbH公司(德国)的Posdziech等[99]开发了一种共电解示范系统,由三个电堆集成,可用来生产合成气(图13)。该系统可以采用不同比例的H2O/CO2混合气作为原料,在输入功率为10 kW的条件下,合成气的最大产率为4 Nm3∙h-1。2020年,奥胡斯大学(丹麦)的研究人员报道,已将SOEC系统与下游合成工艺成功结合,并成功在10 Nm3∙h-1的试验装置中将生物沼气升级为管道质量甲烷[100]。这套系统目前已持续运行超过2000 h,其中SOEC电堆的耗电量为3.07 kWh∙Nm-3 H2,电能利用效率可达到80%左右[101]。近日,日本新能源产业技术综合开发机构(NEDO)宣布,将在2020—2024年累计投资约45亿日元,用以开发CO2电解与费托合成(F-T)耦合制备液体合成燃料(汽油、柴油、航空燃料等)一体化生产技术[4]。
《图13》

图13 Sunfire GmbH公司开发的共电解示范系统[
除了与费托合成路线耦合制备碳氢燃料之外,近期,人们还开发出利用SOEC电催化反应定向精准合成各种化工原料的工艺,如以N2、CH4、C2H6为原料定向合成NO和C2H4等小分子,如图14 [1]所示。这些小分子原料又可进一步经由催化氧化、聚合或其他化工步骤,得到化肥、合成树脂、合成橡胶等一系列具有高附加值的化工产品。
《图14》
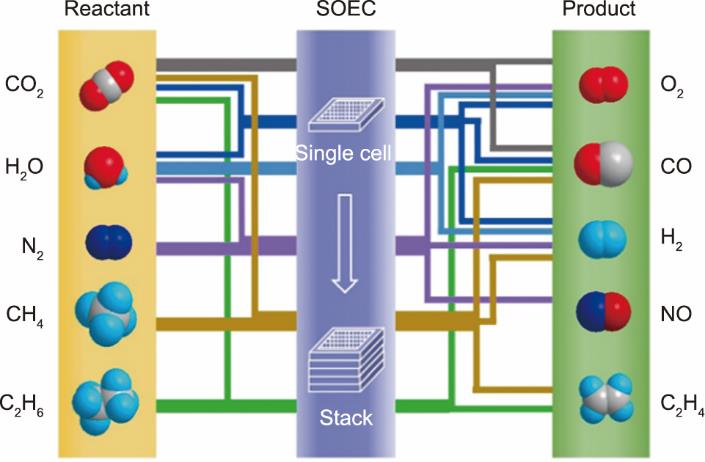
图14 基于SOEC的反应器实现各种化工合成过程的示意图[
一般认为,通过SOEC制备小分子化工产品,在整体效率方面具有独到的优势,这是由于SOEC可以在高电流密度下运行(尤其是对于CO2电解),因而可以实现较高的H2和(或)CO产量[4]。此外,从能源利用的角度来看,SOEC与一些化工生产过程(如甲烷、甲醇和氨的合成)相集成,可以产生许多的优势(图15)[4]。上述这些化工过程均为放热反应,因此在高温条件下运行的SOEC可以有效利用这些过程释放的热量(产生高温水蒸气)进行电解,从而实现能量的最大化利用,形成实质性的协同效应。特别地,对于与合成氨耦合过程而言,在外电场作用下,SOEC单元还可以实现空气中N2/O2的分离,实际上还起到了氧分离膜的作用,从而消除了昂贵的空气分离单元的建设成本[图15(c)]。上述这些合成方法颠覆了传统的化工产品主要来源于石油的制备路线。未来,随着可再生能源发电技术的进一步发展以及可再生电力的降价,这种不依赖石油的SOEC耦合化工制备合成工艺有望成为化工产业的新选择。
《图15》

图15 SOEC耦合化工合成过程的示例。(a)合成甲烷;(b)合成甲醇;(c)合成氨[
《4.3 挑战与未来发展方向》
4.3 挑战与未来发展方向
尽管高温电解技术已经取得了重大进展,但要实现高温CO2电解的大规模应用,还有一些技术领域亟待突破,包括:①开发高活性和稳定性的电解池组件材料;②开发大规模的CO2收集纯化技术;③开发用于高温SOEC电堆的高效热管理系统;④开发出口气体分离纯化技术[2,102]。目前,人们已开展了相关研究,以提升基于SOEC的CO2转换系统的性能[103‒107]。例如,为了处理天然气或煤气化产生的CO2进料气可能含有的H2S等杂质,人们开发了一系列抗硫材料作为SOEC阴极,并开发了各种入口气体预处理系统,以减轻电极中毒现象[2,105]。此外,为了提高转化系统的热效率,人们将放热化学合成过程中释放的热量以潜热或显热的形式加以利用[103]。此外,为了从空气或工厂中获得高度浓缩的CO2,有人提出将碱性化学吸收剂作为大规模CO2储存和释放的中间体[107]。未来,随着这些相关技术的发展和应用,高温CO2电解装置的可靠性和运行稳定性将得到进一步提高。
然而,SOEC技术仍然在经济性方面存在一些问题,包括:①由于电池部件的衰减问题,导致运行和维护成本高;②电解过程耗电量高,导致生产成本高;③由于SOEC部件缺乏标准化制造流程,现有SOEC工厂规模有限,导致建设成本高[1‒2]。为了解决这些问题,建议该领域未来向如下方向发展:
(1)继续开发具有高活性、高稳定性的廉价电池组件材料,以减缓电池材料的衰减,延长电池组件的寿命。此外,还有必要加强开展与SOEC组件在高温原位环境中衰减机制相关的基础研究,由此指导设计更加耐用的电池材料和电堆。
(2)开发具有新型结构(如蜂窝结构微通道电极)的电解池,由此促进高温电解的基元反应步骤(包括气体吸附、电子转移、离子扩散等)动力学进程,并开展单体电解池系统化工程,将其按比例扩大成为电堆和模块,同时确保气密性和操作稳定性。
(3)探索高温电解与可再生能源的耦合模式,包括开发能在可再生能源间歇电力输入下连续稳定运行的SOEC电堆和模块。
上述改进有望显著降低SOEC的运营成本,拓宽其应用场景,并进一步推广SOEC与化学工业耦合,提供实现碳中和的解决方案。
《5、 结论和展望》
5、 结论和展望
基于SOEC的CO2高温电解技术,对于中国实现碳减排、碳达峰和碳中和目标具有重要意义。通过消耗电能,SOEC可以直接将CO2(或CO2/H2O)转化为CO(或碳氢化合物燃料),从而实现CO2的循环利用。CO2高温电解技术目前已在实验室和中试规模取得了很大进展,但该技术的大规模工业应用还有待进一步发展。目前面临的主要挑战包括如何提高在高温、大电流密度下的工作效率和稳定性,如何确保长期耐用性,以及如何推动电解池的系统化和放大,将其扩大成为电堆和模块。为了应对这些挑战,有必要探索开发具有优异电化学性能、高稳定性、低成本的电池组件材料,开发具有新型结构的电池和电堆,以及探索高温电解与可再生能源的耦合模式。在未来,应进一步加强高温电化学领域基础研究,加快先进原位表征手段和模拟分析手段在该领域的应用,以指导开发适用于CO2高温电解过程的SOEC阴极、阳极和电解质材料体系。此外,还应开发高温CO2电解系统的关键配件,包括电源、气体净化系统、气体循环系统和温度控制系统等。与此同时,还应加快开展更多理论模拟和实验研究,来进一步验证可再生能源发电、CO2电解与化工合成过程耦合的经济性和技术可行性,为后续建成大规模CO2资源化利用全产业链奠定理论依据和实验基础。











 京公网安备 11010502051620号
京公网安备 11010502051620号




