

《1、 引言》
1、 引言
人类社会发生翻天覆地的变化,离不开对自然的无穷探索。这种变革性的发展已经从自然观察演变为通过各种工具和前沿方法逐渐实现[1‒2],并逐渐形成涵盖各个学科整体和相互关联的不同规范发展范式[3‒4]。每一次范式转移都是由统治理论内部的基本假设在一定时代内为了适应后续的要求而发生变化导致的,从而产生新的范式[5]。第五范式现在被描述为智能驱动、以知识为中心的研究范式,紧随数据密集型第四范式的转变,紧随实验、理论和计算机模拟范式从第一范式向第三范式的转变[6‒10]。
对第五范式来说,对物理宇宙的探索不仅仅是由智力驱动的密集数据的数学可能领域所投射出来的,而且整个研究过程也涉及人类专业知识的无差别意识过程。基于这些特征,第五范式的应用可以被视为一种认知系统或认知应用[9‒10]。以材料科学的发展为例,第五范式的认知系统是通过经典的螺旋进化过程从原始的早期范式演变而来。在牛顿定律和相对论出现之前,金属和陶瓷等材料在古代就已经被发现和使用。然后,相对论和量子力学的出现使得模拟分子的电子结构成为可能[11‒13]。近年来,人工智能(AI)和机器学习的迅速兴起促进了数据驱动材料设计的研究[14‒18]。因此,通过将相关创新技术加工成越来越大的数据集,可以找到金属和陶瓷等新材料的隐藏特性[19‒22]。特别是当下基于智能驱动的认知材料研究,接过数据密集型材料的接力棒形成了一种新的发展趋势,进一步加快材料科学的探索进程。
目前,第五范式正处于萌芽期,还有很长的路要走。材料领域的智能驱动方法伴随着数据密集型科学研究范式的发展而被逐渐应用到材料创新中。随着成熟的数据密集型科学研究范式在多个领域迅速暴发,有关自动驾驶汽车、计算机视觉和大脑建模等工业和科学等领域的应用技术被广泛开发并逐步实现[23‒27]。以知识为中心的第五范式仍处于蓬勃发展阶段,与之不同的是,数据智能驱动在认知应用中需要打破传统科学计算和数据密集型研究的界限,通过融合和扩展现有技术形成新的生态系统。一些科学家正逐步针对这一需求开展研究,例如,Malitsky等[10]提出的MPI(Spark-message passing interface)集成平台,可用于推动数据密集型应用向智能驱动方向转变;Zubarev和Pitera [9]研究的认知计算,如自然语言处理、知识表征和自动推理,能够促进认知特征面向智能驱动的实现。在材料研究领域,数据智能驱动需要基于不同领域专家知识的整合,以及来自实验观察和理论模拟的大量数据整合,以此推断面向跨学科应用之间的共同属性,设计出更多研究方案以促进材料创新的发展。因此,尽管第五范式的发展任务艰巨,但其应用前景十分广阔。
从数据密集型科学向复合认知计算应用的第五范式的战略转变是一个长期的过程,有许多未知因素。本文通过解析催化材料(https://github.com/ulissigroup/GASpy)[28]中称为Python广义吸附模拟(GASpy)的框架来解决第五范式平台,旨在将人类智慧和高性能计算中的算法和深度学习方法结合起来,以解决数据驱动应用的新领域。本文的其余部分组织如下:第2章提供了对第五范式平台的简要概述和讨论,第3章进一步阐述了平台的性能评估,第4章总结了本文工作。
《2、 第五范式平台》
2、 第五范式平台
在材料研究过程中,实验数据、理论模型和机器学习的协同作用,依赖于不同领域的专家协同分析和处理数据,需要巨大的人类智慧,这些环节均可以通过智能驱动实现。将各个环节的通用结构相结合,实现智能驱动就显得尤为重要。本章介绍一个在催化材料领域使用的第五范式平台,如图1所示。第五范式平台与第三范式和第四范式平台耦合,后两者包括第一范式和第二范式的过程。其中,原始数据来自第一范式的实验观察和第二范式的理论指导,以及第三范式的数值计算,然后可以通过第四范式的机器学习进行智能驱动。结合实验专家和理论专家的工作集成知识,可以对机器学习选择的材料进行第二次筛选,筛选结果被再次反馈到第三范式的数值模拟中。在第三范式中获得的结果仍然可以由第四范式中的数据驱动。然后,通过实验专家和理论专家的知识整合,对预测结果进行再次过滤,再反馈给第三范式进行数值模拟。这些方法产生了第五范式平台。通过智能控制高通量物理模型的计算,不断为机器学习提供样本,以弥补机器学习样本的不足。此外,利用整合到不同领域的知识,机器学习可以代替部分数值计算,解决由于计算资源不足而导致的大量模型耗时的问题。
《图1》

图1 科学中的范式。科学范式的演变已经从简单的第一范式发展到复杂的第五范式。第五范式的核心是以知识为中心和智能驱动,包括从第一范式、第二范式、第三范式到第四范式,分别以实验、理论、模拟和数据驱动过程为标志。
第五范式平台的综合工作源于Tran和Ulissi [28]为材料科学双金属催化剂研究设计的框架,该框架使用机器学习来加速基于密度泛函理论(DFT)的数值计算,该计算由维也纳大学Hafner小组开发的模拟包(VASP)[29]进行,可以推动高性能电催化剂的发现。该平台可以对双金属晶体各稳定低折射率表面的活性位点进行分类,得到成百上千个可能的活性位点。同时,采用基于人工神经网络的替代模型来预测这些位点的催化活性[30]。发现的高活性位点可以进一步用于未来的DFT计算。
《2.1 自动模型构建和验证》
2.1 自动模型构建和验证
智能驱动原始数据提取的能力体现在模型的自动构建中,由第五范式平台验证。可以自动构造有或没有吸附物的更大结构并通过DFT计算对该结构进行验证。由于表面物质的吸附是多相催化不可缺少的过程,在通过评估吸附能来确定催化活性之前,在实验和DFT计算中构建许多结构可能会耗费大量时间。因此,自动化模型构建和验证对于解决问题至关重要。
如图2所示,整个任务计算包括标准模拟原始数据的准备过程,然后进行数值计算。理论模拟所使用的原始数据全部来自Material Project网站,通过定制模块Generate_Gas/Generate_Bulk实现,并将用户信息、任务位置、计算状态和其他属性通过键值对存储,创建名为“Fireworks”“Atoms”“Catalog”和“Adsorption”的集合。
《图2》
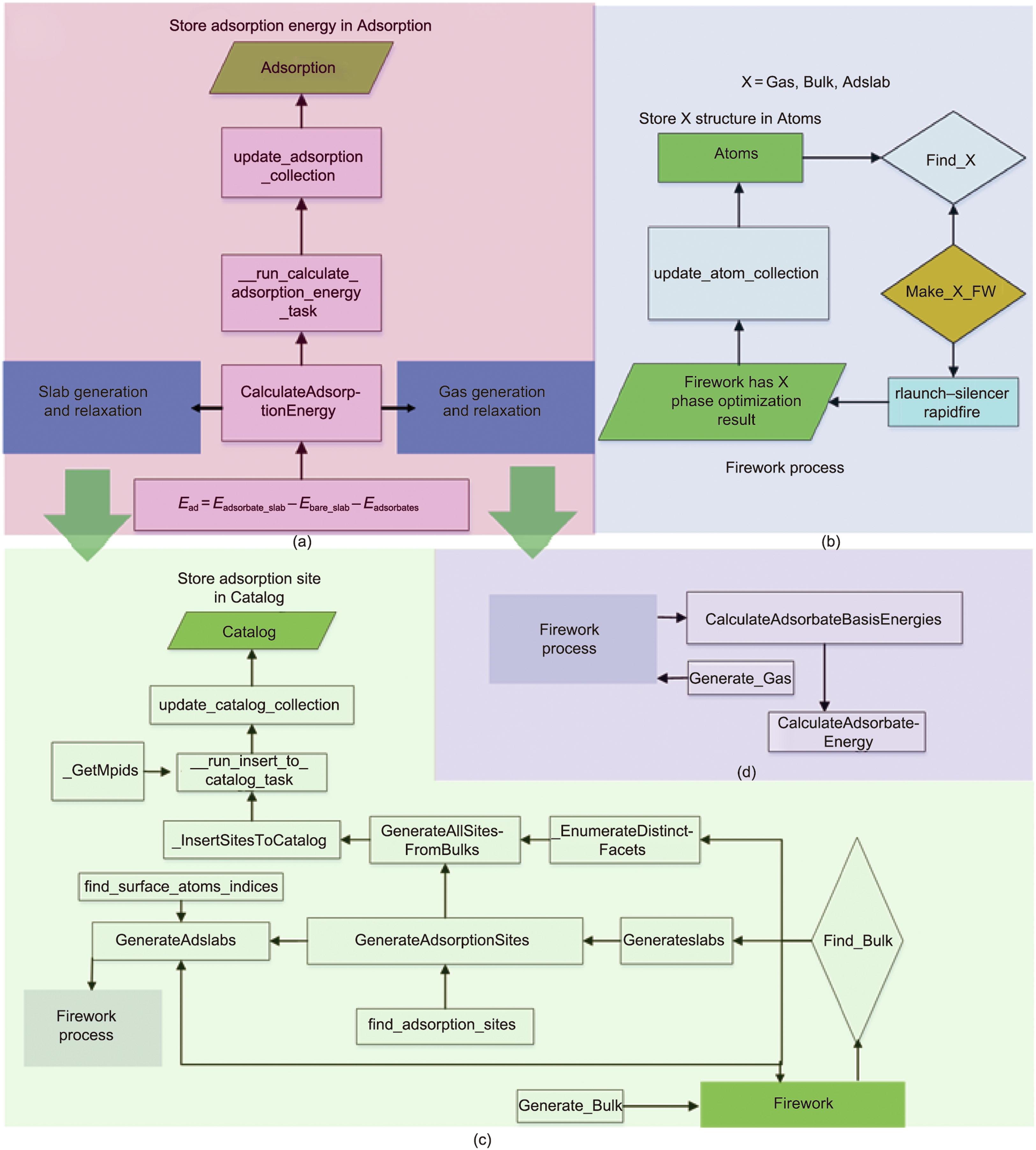
图2 第五范式框架示例。通过原子运算、生成和计算模块,实现了GASpy框架下原始数据提取的智能驱动。(a)该模块的功能是自动计算第五范式平台中气体和板状相的吸附能。(b)该模块用于通过Firework自动创建高通量任务,用于优化有/无吸附质的Gas、Bulk、Adslab。(c)、(d)模块表示部分(a)中描述的Slab生成(c)和Gas生成以及结构弛豫(d)。
然后,FireWorks工作流管理器可以生成图2(a)中任务的松弛计算,以便在图2(b)提交。FireWorks中的结果属性包含“gasphase optimization”作为气体松弛的列表格式,“gasphase optimization”用于批量优化(bulk_relaxation)。属性“status”是“COMPLETED”“RUNNING”“READY”和其他状态(如“FIZZLED”等)的计算状态,由Find_Bulk/Find_Gas函数判断,以将完成的计算过程存储在Atoms集合中,或者生成等待尚未开始的计算的FireWorks任务工作流。
如果Find_Bulk/Find_Gas确定的状态是“COMPLETED”,则把计算结果存储到数据库中,再从Atoms 集合中获取优化后的晶体结构,进行不可约晶面指数枚举(通过EnumerateDistinctFacets函数实现),再根据给定米勒指数,通过扩胞(Atom_operates的函数),枚举晶体表面slab,添加吸附物,从而找到切面的所有吸附位点(由GenerateAdsorptionSites函数实现),如图2(c)和(d)所示。对于所有材料上的指定米勒指数切面吸附位点,可通过由EnumerateDisdinctFacets函数和GenerateAdsorptionSites函数组成的GenerateAllSitesFromBulks函数进行遍历并生成所有吸附位点。所有这些信息都由函数update_catalog_collection 写入 Catalog集合。
对于每个找到吸附位点的切面,通过GenerateAdslabs函数将吸附物添加到吸附位,生成“slab + adsorbate optimization”计算模型(adslab_relaxation)。还可以通过GenerateAdslabs函数去掉吸附物,生成“bare slab optimization”计算模型(bare_slab_relaxation)。然后可以通过FireWorks工作流管理器提交这些计算模型进行计算。
完成后,所有计算结果将通过函数update_atom_collection存储到Adsorption集合中。Find_Adslab函数将通过查找Atoms集合中是否存在相应的计算结果来确定是否应该再发射对应的DFT任务。对于吸附能量 Ead计算,CalculateAdsorptionEnergy函数用于从Atoms集合中提取吸附物能量Eadsorbates、含吸附物的切面能量Eadsorbate_slab和不含吸附物的切面能量Ebare_slab,利用update_adsorption_collection函数将Ead和相关的初始及最终结构等其他信息添加到Adsorption集合中,构成下一步机器学习提取标记指纹的数据集来源。以上过程为整个计算流程的实现。
《2.2 自动指纹构建》
2.2 自动指纹构建
神经网络特征选择的智能驱动性体现在第五范式平台的自动指纹构建中。在该框架中,自动构建的指纹由每个材料吸附模型的所有原子结构转换为卷积神经网络(CNN)数值输入的图形表示[31]。在原子结构信息中,考虑了三种类型的特征,如图3所示,即原子特征(
《图3》
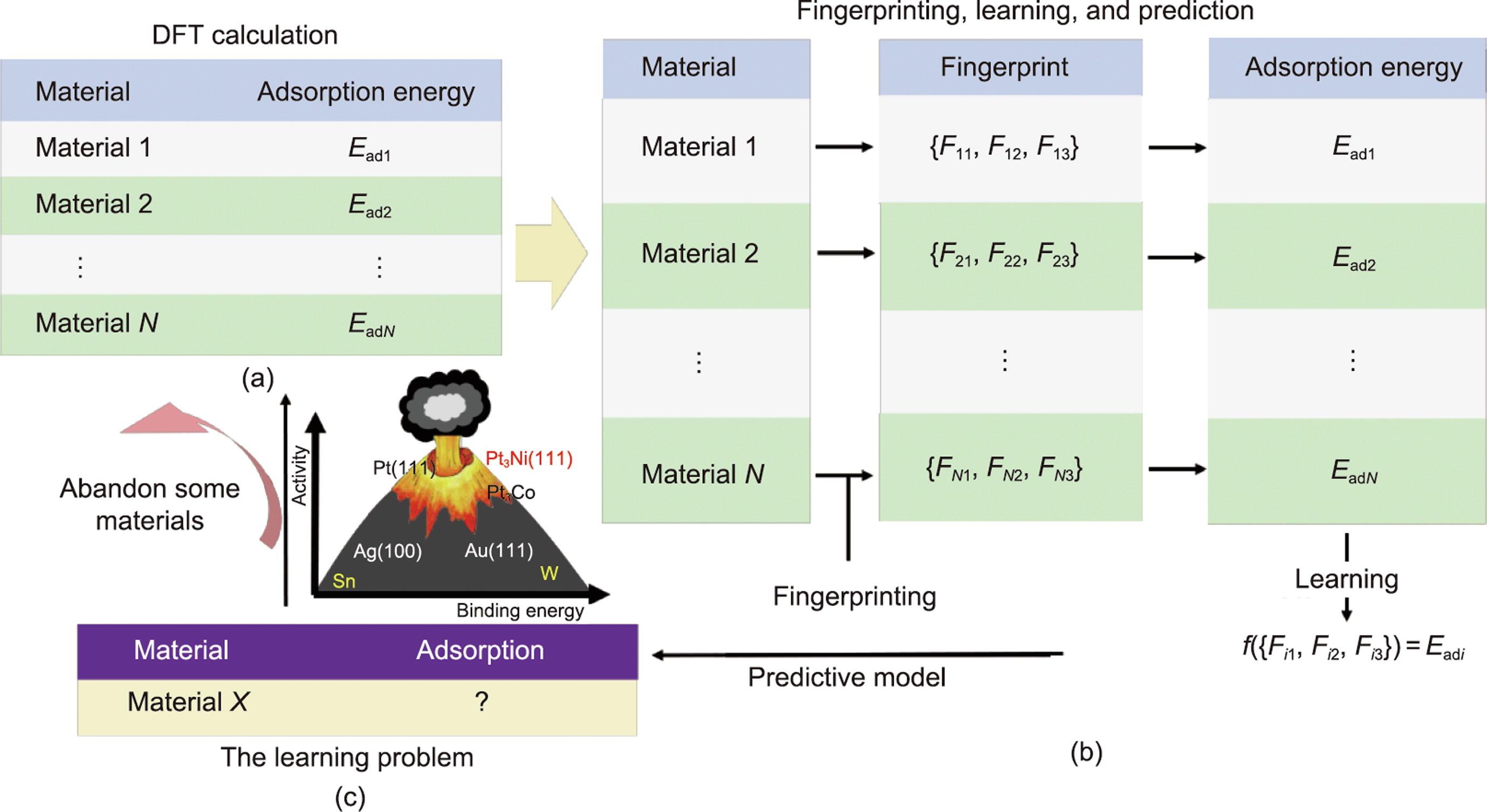
图3 第五范式平台中神经网络特征选择的智能驱动。它是在GASpy框架下通过自动指纹构建来实现的。(a)DFT计算被示意性地视为示例数据集(N是训练示例的数量);(b)通过指纹识别和学习步骤过程,利用预测模型实现自动指纹构建;(c)陈述了学习问题,然后通过比例关系从学习结果中放弃一些材料,并进行进一步的DFT计算筛选。
自动指纹构建过程包括通过DFT计算提取最终结构和吸附能的过程、指纹生成过程、机器学习过程以及学习问题的陈述。GASpy中构建的指纹来源于原始模型,没有DFT计算,也没有DFT计算结果。首先,经过DFT计算,得到初始标记指纹Ead
火山标定关系中的吸附能和催化活性数据来自理论和实验科学家多次尝试的工作,用来作为特征工程中数据清理的标准。以火山标定关系为依据,图3(c)中描述的预测材料将被进一步利用,意味着火山标定关系不匹配的预测吸附能材料将被丢弃。在下一个周期,被利用的候选者将通过DFT再次计算以增加数据集。随着DFT计算的材料类型的增加,数据集的数量在增加,自动化的探索过程使指纹数量不断得到更新。
《2.3 DFT计算和机器学习的理论模型》
2.3 DFT计算和机器学习的理论模型
在第五范式平台中,Kohn-Sham理论与一种集成CNN和高斯过程(GP)的方法[31,35‒37]是DFT和机器学习过程的核心理论模型。因此,简要介绍这些理论模型的细节。
《2.3.1. DFT计算的理论模型》
2.3.1. DFT计算的理论模型
DFT计算是第一性原理计算的典型代表。通过DFT计算获得吸附能是目前研究材料结构的主要手段之一。吸附能计算过程主要涉及通过不断调整原子和电子结构来达到最终能量稳定的结构状态的每个面结构的优化过程。通过基于量子力学近似求解多体薛定谔方程(many-body Schrödinger equation)求解Kohn-Sham方程的DFT方法是该近似解的主要方法之一。
Kohn-Sham方程为:
(1)
(2)
(3)
(4)
给定一个包含K个离子的体系,即三维坐标空间
自洽迭代过程描述如下:
给定一个任意
(5)
式中,occ.代表已占轨道的数量,于是
(6)
式中,H代表波函数ψ的哈密顿量,其能量用ε表示;然后可以获得一个新的电子密度,
(7)
于是
(8)
…
(9)
当
《2.3.2. 机器学习的理论模型》
2.3.2. 机器学习的理论模型
卷积馈送的高斯过程(CFGP)[37]是一种使用网络卷积层的池化输出,为高斯过程回归器提供特征的方法[38]。该方法通过将晶体图卷积神经网络(CGCNN)和高斯过程(GP)相结合,进行训练,产生均值,实现关于以吸附能为代表的材料性能预测。Chen等[39]、Xie和Grossman [40]将CNN应用于晶体的图形表示之上,用来预测各种特性,并由Back等[31]进一步使用Voronoi多面体[32]收集邻居信息的改进方法,用来预测非均相催化剂表面的结合能(如吸附能)。在CFGP方法中,首先训练一个完整的CNN来创建最终的固定网络的权重,卷积层的所有池化输出都用作GP中的特征,再通过使用这些特征来训练GP生成对吸附能的平均和不确定性预测。
在CFGP方法中,晶体结构由晶体图G表示,其中表示晶体中原子之间连接的原子和边由具有原子特征和近邻特征信息的节点编码提供,而CNN构建在无向多重图的顶部[40]。由于晶体图的周期性特征,同一对端节点之间允许存在多条边,每个节点
(10)
式中,
(11)
式中,
(12)
通过优化代价函数
(13)
此时,相应地学习权重W代替目标属性
(14)
式中,
《2.4 机器学习和数值计算之间的迭代》
2.4 机器学习和数值计算之间的迭代
第五范式平台的智能驱动、以知识为中心的本质可以通过机器学习和数值计算之间的迭代被很好地描述,这些迭代由“火山图”的跨学科知识串联起来。这突破了机器学习与数值计算之间人工筛选研究的新材料瓶颈,实现了科学实验与AI的相互促进,如图4(a)所示。实验涉及从Materials Project网站获取原始晶体(或原胞)并存储在数据库中的过程、进行火山标定关系信息比对的过程,以及智能模型的构建以创建大量吸附能计算模型的过程。通过第一性原理计算,将优化后的模型和吸附能数据存储在数据库中,并从中提取指纹以训练合适的机器学习模型。训练后的模型可以使用从尚未经过理论计算的待筛选材料中提取的指纹来预测吸附能,并将结果再次存储在数据库中。通过鲁棒松弛方案,比如对切面最低预测吸附能量的吸附点模型进行智能分析,从中筛选需要进一步开展DFT计算的模型。整个循环如下:①②③④⑤⑥⑦⑧⑨⑩, ④⑤⑥⑦⑧⑨⑩, ..., ④⑤⑥⑦⑧⑨⑩。
《图4》
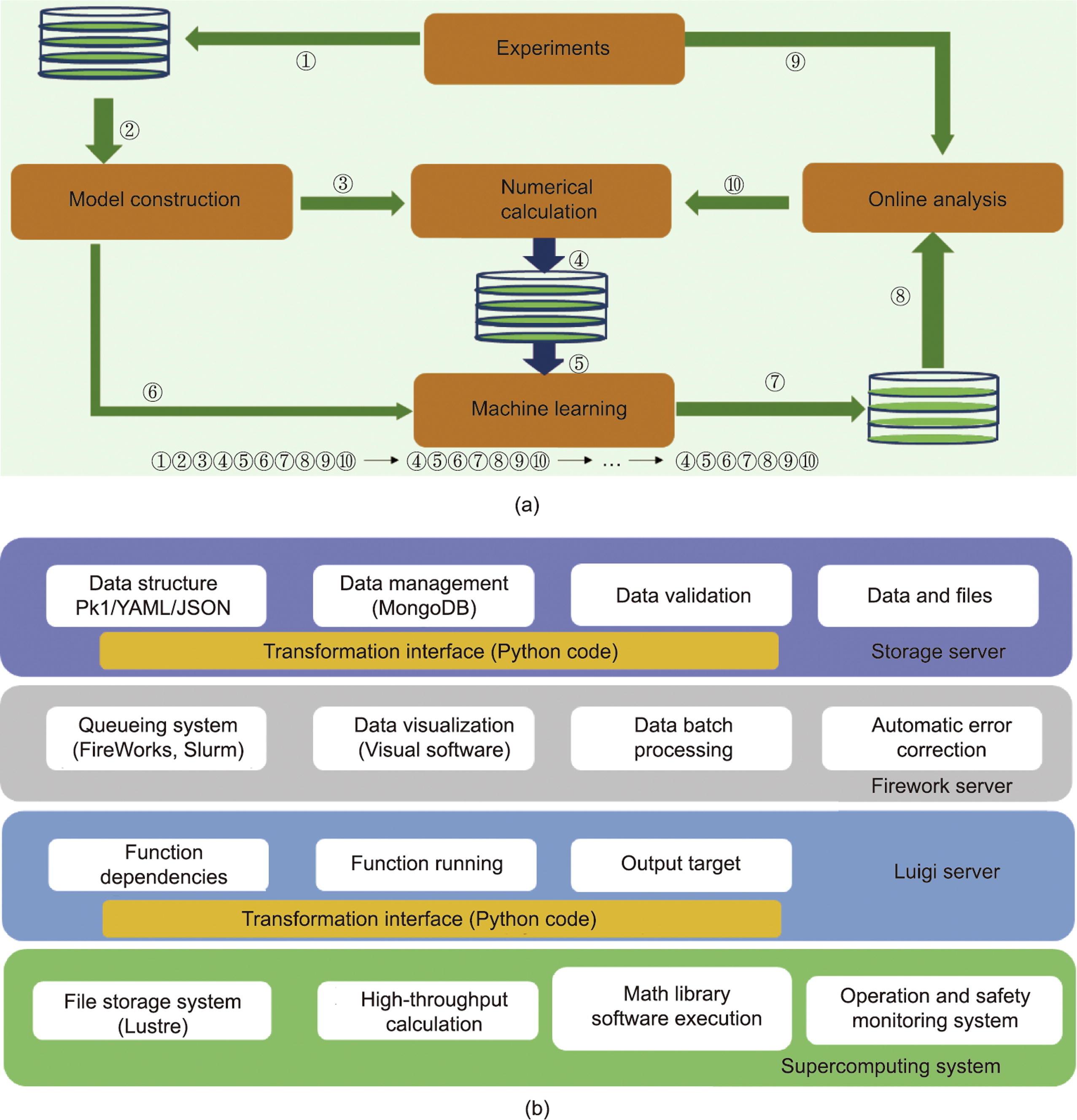
图4 第五范式的架构。(a)重复迭代框架包括第五范式平台中的机器学习和数值计算。步骤①和②以及步骤⑦和⑧分别将实验结果和机器学习结果拉入和拉出数据库。步骤③显示了为从头计算准备的构造模型。步骤④是计算结果的存储过程,步骤⑤和⑥分别是从计算结果和实验结果中提取的指纹。步骤⑨是指通过“火山图”对机器学习结果进行在线分析。”步骤⑩显示了在线分析(筛选)后的剩余模型,这些模型需要进一步的数值计算。(b)服务和功能的实现基于天河一号超级计算机的第五范式平台。GASpy中专用于服务的类型组件包括存储服务器、Firework服务器和Luigi服务器。超级计算系统的基本环境是在软件层面。
当且仅当计算框架中所有材料预测或计算完毕时,机器学习与DFT计算迭代反馈的过程才会停止。机器学习与第一性原理融合的功能在这些步骤中得到了很好的体现。步骤⑤表示数值计算得到的数据集补充了机器学习过程中没有数据集和数据集较少的问题。步骤⑩表明,通过机器学习预测后,再利用火山标定关系可以过滤掉大量数值计算,达到实现材料快速筛选的目的。此外,机器学习的结果可以通过整合实验和理论科学家的知识(跨学科专家的协同作用)的“火山图”进行智能分析,形成以知识为中心的智能驱动的第五范式。
《2.5 信息科学工具》
2.5 信息科学工具
第五范式的框架是通过使用各种Python包构建的,如Python Materials Genomics(pymatgen)、自动模拟环境(ASE)、FireWorks、Luigi和MongoDB [42‒45]。pymatgen是Python支持的用于高通量材料计算的功能强大的程序包之一。该程序包标准化了运行高吞吐量计算之前所需的初始化设置,并提供了计算生成的数据的过程分析。ASE旨在建立、引导和分析原子模拟。FireWorks的功能是对运行在高性能计算集群上的高吞吐量计算工作流进行作业管理。Luigi可以用来构建复杂的批处理作业管道,处理依赖关系,并进行工作流管理。MongoDB是用C++语言编写的,用于实时数据存储,可以共同满足Java-Script Object Notation数据交换格式。
如图4(b)所示,在基于Lustre文件系统的天河超级计算机上开展数据密集型DFT计算工作[46],可以通过运行部署在集群上的安全监控系统及服务实现高通量任务的管理与调度。针对该模型设计两类服务:Luigi服务和FireWorks联合MongoDB的服务。Luigi通过管理和解析依赖(函数依赖、运行和输出目标),构建各种物理模型,再通过FireWorks对任务管理系统进行配置和计算,通过超级计算机[47]中的Slurm资源管理系統对这些任务进行批处理提交。综上,基于天河超级计算机的高通量材料计算环境能够灵活管理和运行单个作业,最终实现高通量任务被持续提交与执行的过程。
《3、 性能评估》
3、 性能评估
为了说明第五范式平台在催化材料筛选中的性能,本研究进行了比较测试,以解释机器学习过程如何加速数值计算,以及数值计算过程如何为机器学习迭代提供可训练的样本。在本文中,没有在每个模型的学习周期中使用包含在线DFT计算过程的更新数据集,而是使用DFT计算数据集提取相应的指纹进行研究。由于目标预测与DFT计算的结构没有直接关系,而是与从初始结构中提取的指纹有关,因此没有进行任何模拟处理。本文研究团队认为这不会影响对平台的评价。
准备的测试交叉验证过程的数据集来自Github(https://github.com/ulissigroup/uncertainty_benchmarking)。由H、CO、OH、O和N五个吸附物组成,其中主要数据集来自前两个吸附物(21 269和18 437)。采用CFGP的方法创建模型,超参数采用第二章用到的设置,通过
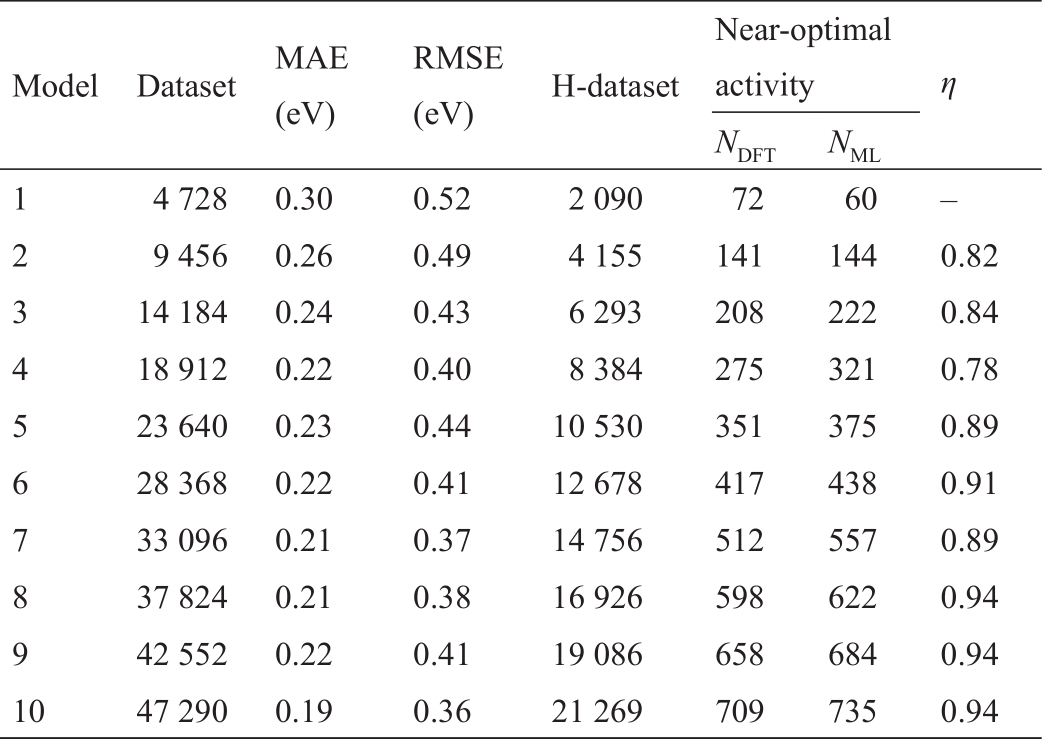
机器学习和数值计算之间相互反馈实现的是DFT计算提供的可训练样本可以补充机器学习迭代。在该平台中,一旦发生迭代,就确定了包含目标特征的数据集,这意味着确定了相应迭代的机器学习模型。此外,作为第五范式平台的典型案例,每个迭代过程的性能比较都是从相同数据生成条件下的模型比较中得出的。根据表1所示,为评估高通量材料计算在智能驱动过程中反馈迭代的机器学习性能,构建了10组数据集。首先将整个数据集随机打乱并拆分为10个模型,将总数据集的10%作为第一个模型数据集,然后递增直到将总数据集的100%作为第十个模型数据集,形成10个模型对应的数据集。前一个模型的数据集包含在下一个模型的数据集中。对于交叉验证过程,每个模型的训练/验证/测试比为64/16/20,如文献[37]所述将所有单金属添加到训练集中。交叉验证及其结果列于表1和图5中,图5(a)为训练和测试数据集的相关系数(
(15)
式中,
《表1》
表1 机器学习模型性能评估
《图5》
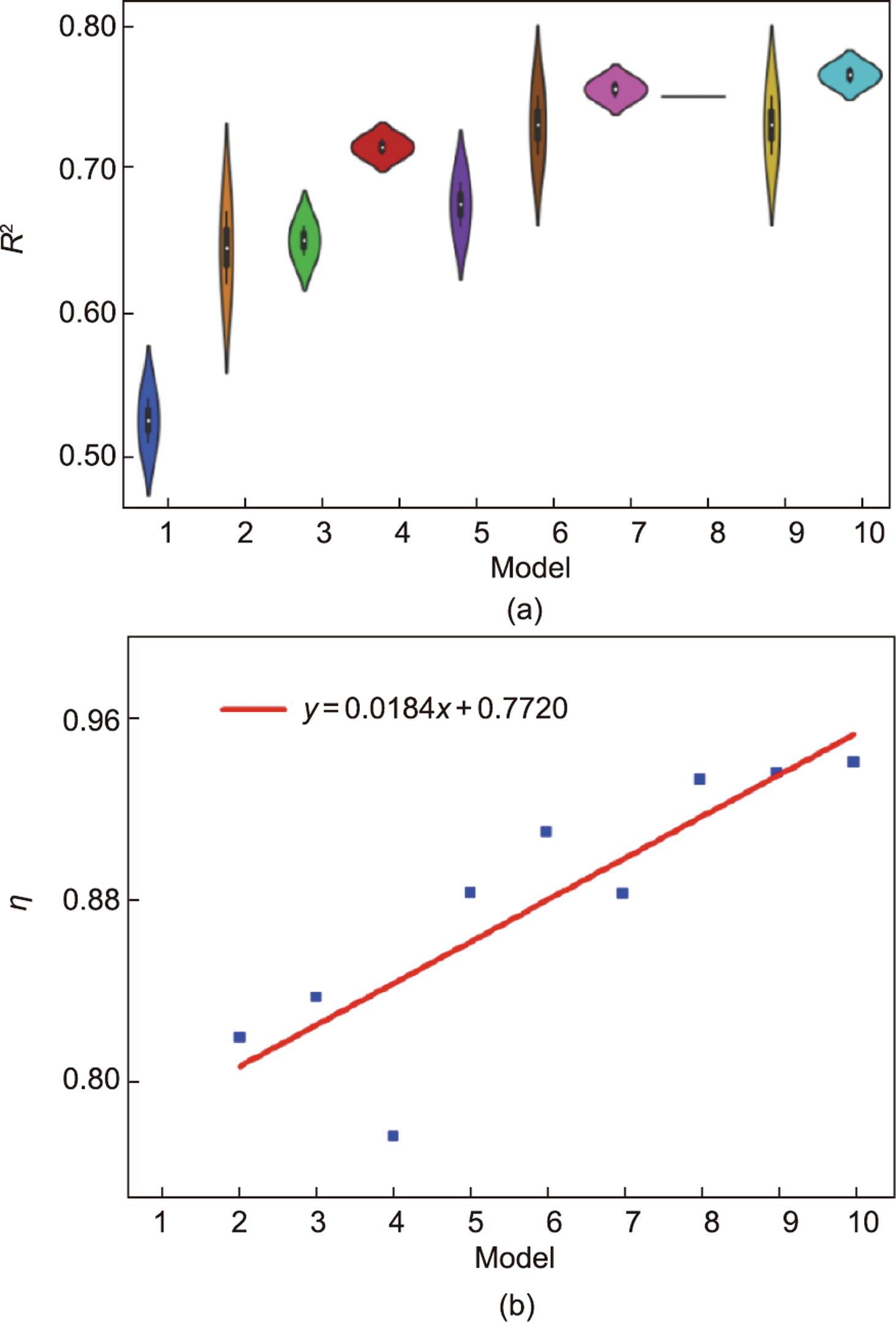
图5 第五范式平台下学习模式的绩效指标评价。(a)10个模型中验证和测试过程的R2相关系数。(b)所有模型的
数据集指每个模型的数据集总数,评价指标包含MAE和RMSE及η。通过DFT计算和机器学习预测验证火山标定关系中H吸附能接近最优范围的切面数量,分别以NDFT和NML为代表,η用于评估模型性能变化的趋势。
为了说明机器学习和数值计算之间相互反馈的实现(例如,机器学习解决了数值计算中计算资源不足导致的大量模型耗时问题,而数值计算过程提供了机器学习训练样本),本文准备了三种类型的预测案例来了解上述训练和验证模型的性能。在预测过程中使用的数据集来自Tran和Ulissi [28]的工作,其中包含22 675个H吸附DFT结果。事实上,该数据集已经涵盖了上面提到的21 269 H数据集的大部分。然而,这与重复数据集无关,因为本研究的目标是比较在大小不同的样本上生成的机器学习模型的性能,并找出大小不同的预测样本下机器学习的加速行为。此外,要预测的与数据集对应的材料结构并不取决于是否进行了模拟计算。因此,该机器学习预测数据集取自DFT计算数据集的决策不会影响对智能驱动过程的整体评估。
就高通量材料计算在智能驱动过程中的反馈迭代特点而言,每个周期(第一个周期除外)进行的DFT计算均从机器学习结果中获得。表2列出了三种方法:累加异测保守丢弃、累加异测和同值异测方法。累加异测方法是指上面形成的model 1到model 10的机器学习模型对应的总预测数据集的10%到100%增量数据集。此外,整个预测数据集也可以在每个周期中保持相同,正如同值异测方法所定义的那样。对于累加异测保守丢弃方法,意味着机器学习在最优范围内预测的模型在下一次模型预测中被丢弃。过程如下:从model 1开始,在预测的整个22 675个模型中,发现机器学习预测的4960个模型是命中的。当使用模型2进行预测时,机器学习预测的模型将从预测的22 675个模型中剔除4960个模型,只剩下17 715个模型(22 675 - 4960 = 17 715)。然后,模型2发现,机器学习预测的860个模型被命中,并提供另一个简化样本16 855(17 715 - 860 = 168 55)用于模型3的预测。在这10个模型的预测完成之前,这种波动不会结束。请注意,NHits应该等于NML,但是样本中的某些材料必须排除在接近最优的活动过程之外。
《表2》
表2 接近最优范围内的三种预测及所有模型性能

表2列出了三种方法在最优范围内的结果。在累加异测保守丢弃方法中,由于前一个模型预测的NML从下一个模型的预测样本中被扣除(model 1除外),所以从model 1到model 10的NDFT、NML和NHits也相应减少。在累加异测方法中,随着预测样本的增加,NDFT和NML逐渐扩展。在同值异测方法中,NML在4177和4556之间波动,而NDFT保持不变。因此推断,这是由机器学习模型的不同精度引起的,模型中的数据集总体呈现越多,NML被预测命中得越多。从加速的角度来看,累加异测保守丢弃方法可以保证在上一轮中被预测过的数据集不会在下一轮中被再次预测,而其他两种方法在每轮预测中涉及数据集的重复预测。因此,理想的累加异测保守丢弃方法保证了所有数据集仅被预测一次,因此每轮迭代过程中不重复的数据集能够提供更快的机器学习过程。
为了评估这些方法在加速DFT计算方面的差异,在图6中比较了NML取代的NDFT的数量,以及NML/NDFT的值。机器学习替代DFT计算的定义如下:
(16)
《图6》
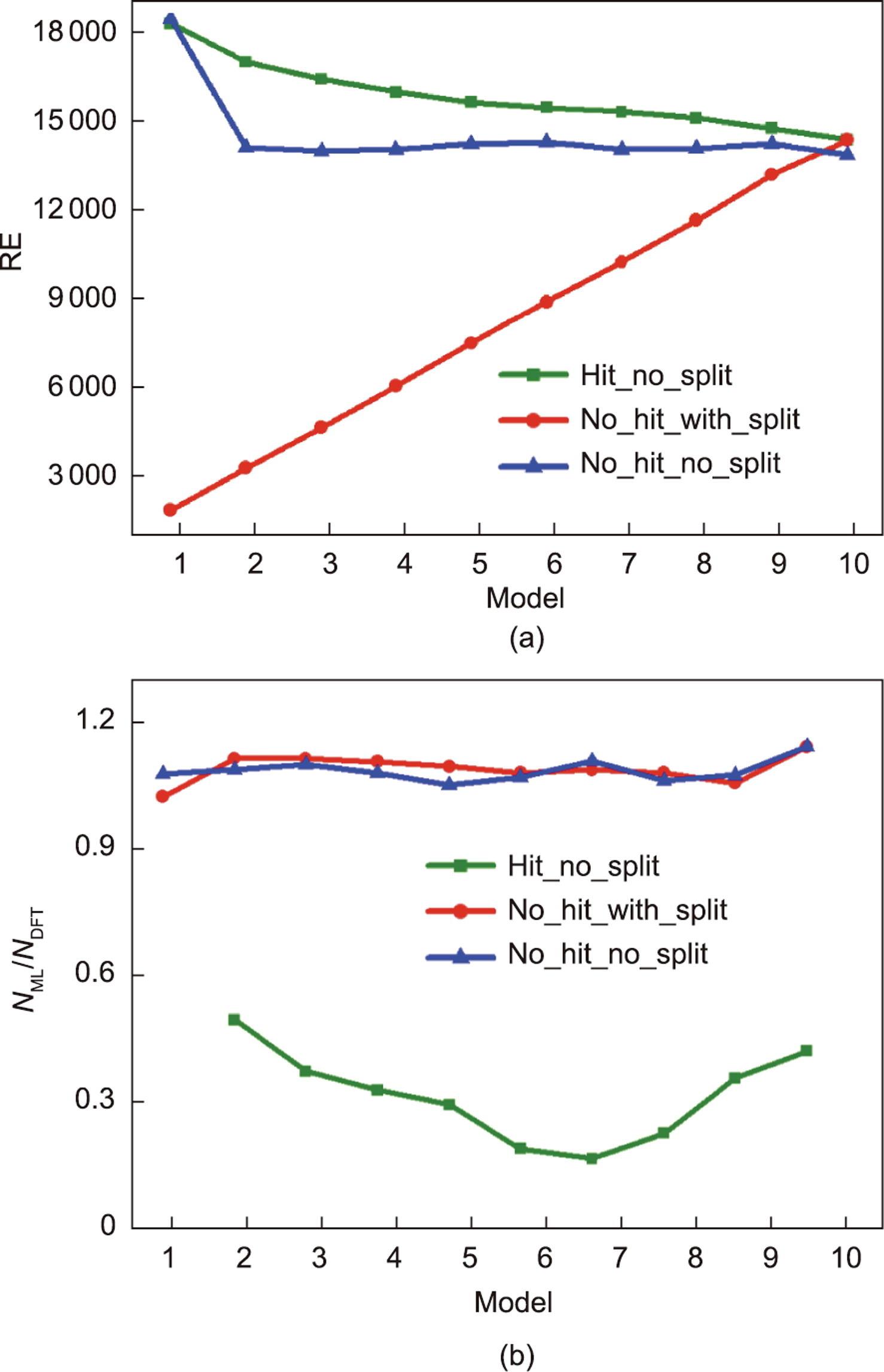
图6 在第五范式平台中构建的所有模型的预测性能。上图是DFT计算(NDFT)的数量替换为机器学习预测(NML)的数量。下图是预测过程中不同模型的NML/NDFT在接近最优范围内的变化。在Hit_no_split方法中,model 1由于其对其他模型的基线函数而被放弃。
式中,RE和
在图6(b)中,通过比较NML/NDFT,可以从另一个角度反映每个模型的性能。理想的NML/NDFT值应该均为1。在累加异测和同值异测方法中,NML/NDFT略微增大到接近1,表明两种方法的预测行为相似,适合加速DFT计算。而在累加异测保守丢弃方法中,model 1作为参考基准不考虑其性能,NML/NDFT值从model 2逐渐减小到model 7,然后在其余模型中逐渐增加,且所有模型均低于0.5。一方面,这些较小值的浮动是由机器学习模型的精度变化引起的;另一方面,随着预测样本命中数的减少,在下一个模型中可以命中的NML逐渐减少。此外,对于累加异测保守丢弃方法,每轮会去掉上一轮的命中数,在下一个模型中可以命中的NML逐渐减少。该方法由于不涉及重复命中材料在其他迭代中被再次命中的情况,因此虽然该方法在速度方面的优势更加明显,但并不适合利用NML/NDFT指标对精度进行评估。
另外,由于机器学习模型本身在交叉验证小样本的扩展过程中呈现出不良拟合逐渐减少的特点,故在model 2到model 10的预测过程中会出现一定程度的精度损失。例如,预测的机器学习数据集本应被命中但未被命中,或者数据集不应被命中但被命中,导致命中数据丢失或未命中数据在下一个模型的数据集中增加。预测样本量不够大,导致机器学习模型的欠拟合或过拟合。累加异测保守丢弃方法具有替代更多DFT计算的优势,虽然不适合用NML/NDFT指标进行精度的评估,但这并不代表累加异测保守丢弃方法不适用于第五范式平台。当预测模型足够好且数据集足够大时,该方法能够减少数据的重复预测过程,同时保持结果的可靠性,以加速机器学习的优势,实现在高精度下加速材料的筛选效果。
基于这三种方法的结果,利用机器学习预测相对于DFT计算的精度损失来评估第五范式平台的性能。精度损失可以定义为:
(17)
式中,L为精度损失。鉴于累加异测和同值异测方法具有相对合适的预测性能,因此只考虑这两种方法的精度损失。如图7所示,在探索未知世界的过程中,科学实验、理论计算和机器学习的相互验证过程代表了材料科学研究范式的准确性。对于累加异测方法,虽然model 1的准确率损失最低,但数据集很小导致机器学习取代DFT的规模非常小,所以速度不高。model 9机器学习取代DFT的规模非常大,且机器学习与DFT计算数量趋向一致,因此精度损失最低。对于同值异测方法,model 5机器学习取代DFT的规模非常大,且机器学习与DFT计算数量趋向一致,因此精度损失最低。因此,随着数据集的扩大,机器学习将继续取代DFT计算,并且会有不同程度的精度损失。
《图7》
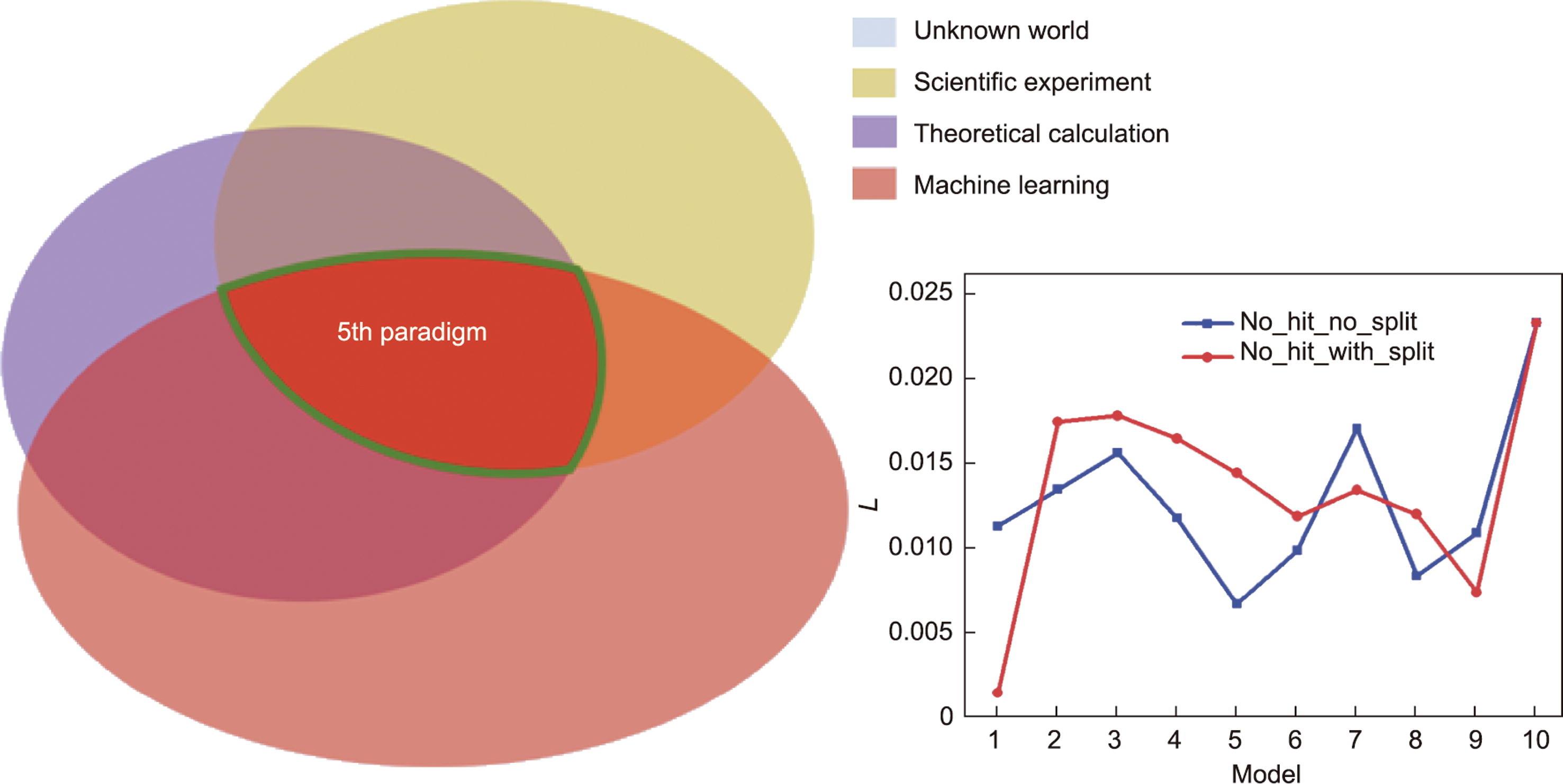
图7 第五范式的准确性。在探索未知世界的过程中,科学实验、理论计算和机器学习的相互验证过程代表了第五范式的准确性。在第五范式平台中构建了所有模型的机器学习和DFT计算之间的No_hit_No_split和No_hit_with_split方法的精度损失(L)。
本研究认为,第五范式的精度损失与机器学习、理论计算和火山标定关系反馈的实验样本量有关,这正是第五范式在精度方面以知识为中心的特征。如图7所示,准确的第五范式应该使机器学习、理论计算和科学实验在共同探索未知世界时呈现一致的结果。虽然这个标准要求很高,但始终是人类对未知世界不断探索以期达到的高度。
《4、 第五范式平台的讨论》
4、 第五范式平台的讨论
模型的自动构建、指纹的自动提取、密集数据与DFT计算的智能耦合以及“火山图”的机器学习构成了第五范式平台的架构。在智能驱动的框架下,充分利用当前各种信息工具和方法的发展,有效地减少了传统模型构建和计算的工作量,大大简化和改进了材料研究中极其繁琐和具有挑战性的工作。
该框架面临的挑战之一是在第五范式中实现的有限应用领域。这是因为第五范式最典型的特征是智能驱动,需要跨学科专家协同进行深入研究。例如,在本工作介绍的材料科学中,需要智能驱动实验专家和理论专家的高效协同,这可以通过“火山图”过滤机器学习结果来实现。对于一些高通量的跨学科工作,在设计类似的第五范式框架之前,最好首先考虑利用适当的方法来量化不同应用领域的专家之间的协作工作。
此外,机器学习模型的鲁棒性和泛化性又与对应的数据集规模有关。缺乏大的数据集将导致训练模型较差的泛化能力,因此需要积累更多的有效数据集才能实现高精度的机器学习模型预测。有效数据集的来源依赖于更多DFT计算产生的结果,比如,目前Facebook AI Research研究团队和卡内基梅隆大学化学工程系联合研发的Open Catalyst 2020 [49]数据集项目正在持续进行相关数据集更新。因此,智能驱动过程中机器学习对于DFT计算产生的有效数据集规模及相关的准确度对量化精度过程非常重要。最后,利用第五范式实现对未知世界探索的准确性受到机器学习、理论计算和科学实验的影响。高精度的第五范式倾向于在其合理的发现、推导和判断范围内,通过三种合作从未知世界中探索同一客观事物。因此,对第五范式案例的剖析可以极大地促进材料科学第五范式在未来的发展。
《5、 结论》
5、 结论
在本研究中,讨论了因人工智能带来的繁荣而出现的最新范式的科学解释。然后,以第五范式平台为典型案例进行了详细的讨论。该平台符合一个具体而明确的框架,能够促进材料科学的发展。跨学科的知识和智能驱动的特征是第五范式的关键,这可以在包括自动模型构建和验证、自动指纹构建以及机器学习和理论计算之间的理论模型和重复迭代的工作中解决。还详细讨论了构建框架所需的信息学工具。最后,进行了测试和比较,以展示在第五范式案例的框架下,人工智能和数值计算之间的相互作用如何有意义地相互促进、减少数值计算,并在相互反馈过程中创建更多可训练的样本。数值计算和机器学习模型以及技术的管理使第五范式平台更具可解释性。
随着数据集的扩大,一方面,机器学习取代的DFT计算越多,材料的筛选就越快。另一方面,最终机器学习预测的候选材料数量与DFT计算的候选材料数量越一致,机器学习的预测就越准确。这种最小精度损失的辨别,代表了科学第五范式下材料研究的精准探索前提,即要求在利用机器学习、理论计算、科学实验共同探索未知世界时,得到一致的结果。
虽然本文为催化材料领域中所代表的第五范式平台提供了科学的解释,但也要承认还有更多的东西需要讨论。跨领域第五范式的整体发展仍然面临着需跨学科专家之间的协同作用和数据驱动学科中数据需求的急剧增长的挑战。尽管面临这些挑战,但可以预见,在各方的共同努力下,人工智能技术与传统学科的结合将不断深化,使每个模拟和计算环节具有更高的智能和自动化特征,最终作为一个平台去运行,从而提高传统科学计算的效率,推动材料研究向更加智能和高精度的方向发展。未来,对第五范式平台的关注可以为第五范式在其他领域的应用铺平道路。











 京公网安备 11010502051620号
京公网安备 11010502051620号




