
《1. Introduction》
1. Introduction
In 1986, Murry et al. [1] first described ischemic preconditioning (IPC), in which brief episodes of ischemia/reperfusion (I/R) preceded an injurious I/R resulting in reduced myocardial infarct size in a canine model. The phenomenon of IPC has since been successfully demonstrated in multiple animal species including dogs, rats, pigs, rabbits, and mice, as well as confirmed in human patients [2−7]; reviewed in Ref. [8]. Marber et al. [9] reported a second window of protection (late preconditioning), which develops 12−24 h after the initial preconditioning stimulus. Unlike early IPC, which is powerful but short-lived, late IPC remains protective for 24−72 h, although the magnitude of the protection is less over time [3]. Repeated preconditioning episodes of the same type are neither additive nor cumulative [10].
In 1993, Przyklenk et al. [11] demonstrated that in the canine heart, a preconditioning stimulus applied to the vascular bed supplied by the circumflex branch reduced infarct size in the area of the myocardium supplied by the left anterior descending coronary artery. Subsequently, it was demonstrated by McClanahan et al. [12] that cardioprotection could even be achieved by preconditioning ischemic stimuli in distant organ sites, such as the kidney. Remote ischemic preconditioning (RIPC) has been demonstrated to elicit cardioprotection following ischemia of kidneys, intestine, limbs, liver, skeletal muscle, and brain (reviewed in Ref. [13]). While the release of diffusible factors from ischemic preconditioned tissues/organs is a key aspect of RIPC [14−18], it is also evident that neural connections to the preconditioned limb or tissue are required. For instance, it has been shown in most [19] but not all [20] animal models that blocking sympathetic transmission using hexamethonium abrogates RIPC’s protective effect. In a mouse hindlimb model, it has been demonstrated that either occlusion of the femoral vein or transection of the femoral and sciatic nerves likewise abolishes the cardioprotection observed after RIPC, indicating the requirement for both humoral and neural pathways [21]. RIPC has been shown to improve short- and long-term clinical outcomes when applied prior to emergency or elective percutaneous intervention [22], coronary artery bypass grafting [23], and valve replacement [24]. Many of the detailed mechanistic aspects of RIPC are shared with IPC (see below). Although IPC and RIPC have emerged as powerful methods of ameliorating I/R injury to the myocardium, their use as clinical cardioprotective strategies to attenuate the pathophysiological consequences of I/R injury (i.e., infarction and ventricular dysfunction) is limited by the inability to predict the onset of clinical ischemia.
As a direct result of considering this limitation, Zhao et al. [25] demonstrated that rapid sequential intermittent interruption of coronary blood flow during the early moments of reperfusion after ischemia attenuates I/R injury. This phenomenon, termed postconditioning, is extremely valuable in that it supports the efficacious use of repetitive ischemia at a clinically relevant time point (e.g., at reperfusion) [26]. The more recent findings in this field [27−30] have combined postconditioning and RIPC concepts by demonstrating that ischemia of a remote site (such as a limb) can elicit cardioprotection at reperfusion. A related phenomenon, perconditioning, results from administering a cardioprotective stimulus during the I/R injury [31,32]. Remote perconditioning involves using a conditioning stimulus at a remote site during myocardial infarction (MI), and has been shown to be protective and to have clinical value [24]. However, all of these cardioprotective strategies have a limitation in that they require the use of an ischemic stimulus to an organ; this is often not clinically desirable. Furthermore, clinical trials of such approaches have been disappointing [33], and/or come with significant limitations and risks that prevent their use [34,35]. The clinical trials of remote ischemic conditioning have some limitations, mainly as a result of including too few subjects, and of confounding variables inherent within the patient population. The patients involved are of heterogeneous age and overall health; many have co-morbidities that are known to reduce the efficacy of cardioprotection, and are administered drugs that mask or block cardioprotection [8]. Furthermore, factors such as collateral circulation and spontaneous or very early clinical reperfusion can lead to small infarcts that do not benefit from adjunctive therapy [13].
Even though recent results with limb ischemia have supported cardioprotection in humans [36], this approach can be difficult to tolerate and cannot be employed in some patients, including the morbidly obese, or patients who have had axial lymphadenectomy. An alternative would be to achieve cardioprotection using a non- or minimally-invasive technique, such as a remote non-ischemic stimulus. Cardioprotection has been shown to result from non-ischemic stimuli including nerve stimulation (spinal, vagal, femoral), acupuncture and electroacupuncture, skin incision, and the chemical or electrical treatment of skin [37−43]. To the extent that some of these modalities are less traumatic yet efficacious, they are being studied as potential therapies.
《2. IPC, RIPC, and cardiac conditioning》
2. IPC, RIPC, and cardiac conditioning
Over the past 25 years, we have learned a great deal about the detailed mechanisms that underlie cardioprotection in general, and IPC, RIPC, and related phenomena specifically. Mechanistic studies usually focus upon initiators, mediators, and effectors of protection, though over the years, our insights have blurred some of these distinctions.
Clearly, understanding the mechanism of cardioprotection after IPC, RIPC, and postconditioning has the potential to elucidate potential therapeutic targets that could be used to initiate or intensify cardioprotection. These targets could be useful in preconditioning in certain situations, including preventing/reducing perisurgical MI and treating patients at risk for MI [5,44,45]. Alternatively, understanding the mechanism may be the key to discovering more clinically applicable ways to initiate perconditioning and/or postconditioning in the clinical setting. Generally speaking, similar—yet not identical—mechanisms underlie the mediator and effector aspects of diverse protective stimuli including IPC, RIPC, preconditioning, postconditioning, and perconditioning [46]. The most diverse aspect of cardioprotective phenomena seems to be in the initiating phase, but this may simply represent the point of stimulation in a more general convergent mechanism. There are some distinct mechanistic differences and specific pathways that have been shown not to be necessary for certain types of cardioprotective stimuli. Combinatorial studies (use of multiple stimuli) have sometimes shown additive effects supporting mechanistic differences [47,48]. However, other combinations have not shown such effects, supporting a similarity or at least an overlap of mechanism. In this paper, we compare and contrast some of these data, extract some meaning, and add to what is known in a way that may point to more efficacious and practical types of cardioprotection to invoke in a clinical setting.
Some studies of combinatorial stimuli are shown as follows. In recent years, there have been several studies combining conditioning stimuli in order to determine if there is overlapping, additive, or synergistic protection. Several groups have studied the combinatorial effects of postconditioning and perconditioning. The rationale for such studies is that the reduction in infarct size by either stimulus is often less than that of preconditioning. Given the greater clinical relevance of these stimuli, any additivity would be of interest. Xin et al. [47] first studied combinatorial effects in 2010. They found that postconditioning and perconditioning, while each less protective than preconditioning, additively mimicked the extent of protection afforded by preconditioning. Furthermore, they found that combining the effects of preconditioning with those of the postconditioning plus perconditioning combination resulted in no further protection, suggesting that preconditioning and the postconditioning/perconditioning combination have similar overlapping mechanisms. Studies of kinase activity showed the activation of Akt and ERK1/2 phosphorylation by all stimuli, but that the extent of activation was highest in IPC and was mimicked by the postconditioning plus perconditioning combination. In each instance, protection was blocked by PI3K or ERK1/2 inhibitors. Xin et al. concluded that the stimuli, which differ in the timing of initiation, work through the same general mechanisms and that it is the extent of kinase activation that confers the extent of protection in mice. This conclusion is consistent with mechanistic studies of these phenomena, and with a study by the same group showing that repeated remote postconditioning stimuli increase the extent of cardioprotection [49]. Interestingly, Tamareille et al. [30] found that IPC and RIPC act additively to reduce infarct size, but that, in addition to the activation of reperfusion injury salvage kinase (RISK) pathways, the addition of RIPC resulted in increased levels of phospho-Stat3. They concluded that the mechanisms of IPC and RIPC were overlapping but that they were somewhat different. However, it must be noted that a requirement for Stat3 has independently been established for IPC [50,51]. A study by Kloner’s group showed that perconditioning and remote postconditioning were not effective in the rat model used, obviating the question of the combination in that case [52]. The recent remote ischemic postconditioning (RIPOST) clinical trial showed that remote perconditioning and postconditioning were both effective, but that the combination did not produce a significant additive effect (reduction in creatinine kinase-MB (CK-MB) AUC from 31% to 29%, P>0.05) [48]. Thus, the evidence of combinatorial studies in animal models and in humans supports the concept that overall, protective pathways work by overlapping if not similar mechanisms. Because of the growing recognition of this concept in the field, the term “conditioning” is coming into vogue as a general term to refer to these types of effects.
《3. Novel methods of initiating cardiac conditioning》
3. Novel methods of initiating cardiac conditioning
In addition to cardiac ischemia, which initiates IPC, and ischemia elsewhere, including limb ischemia, which initiates RIPC to the heart, several non-ischemic stimuli have been studied as initiators of cardiac conditioning, notably electroacupuncture (EA), skin incision, and the pharmacological activation of skin nociception. We review these stimuli here due to their potential clinical relevance, and for what they can tell us about the mechanism of cardiac conditioning. We primarily discuss the effects of EA, spinal stimulation, vagal stimulation, and skin nociceptor activation, all of which are able to activate cardiac conditioning. This work was presaged by three papers from the late 1980s and early 1990s that showed the adaptive effects of EA and vagal stimulation upon the heart [53−55]. In particular, the paper published in 1991 by Meerson et al. showed that EA of an isolated heart reduced necrosis. Subsequently, several investigators observed a role for nerves and neuropeptides in conditioning phenomena. The peptide bradykinin is well known to be required for ischemic pre- and post-conditioning [56,57], and Erşahin et al. and Shoemaker et al. first showed its involvement in RIPC [58,59]. Bradykinin is involved in sensory nerve signaling, and cardiomyocytes have the BKR2 receptor for bradykinin. In the rat model, bradykinin in the coronary circulation is degraded quickly by angiotensin converting enzyme (ACE) and by aminopeptidase P. Erşahin et al. [58] showed that apstatin, an aminopeptidase P inhibitor, is cardioprotective against I/R injury in an isolated rat heart model, supporting the idea that the local action of bradykinin is protective, but not ruling out effects of systemic bradykinin.
The Vatners’s group later showed that innervation and specifically the α1-AR is required for the late phase of IPC in a porcine model, but that the early phase of IPC is independent of this pathway [60]. RIPC is known to require intact innervation from the ischemic bed; while there is evidence for hormonal mediators, it appears that neural signals are critical as well [61]. In addition to neural involvement in ischemic stimuli, all known forms of non-traumatic/non-ischemic remote conditioning involve the neural system at some level. EA is now thought to work through the stimulation of skin sensory or other innervation. Vagal and spinal stimulation invoke the direct stimulation of nerves. We have shown that skin nociception-induced conditioning (NIC), whether by skin incision (remote preconditioning of trauma, or RPCT), or chemical nociceptor stimulation, involves the stimulation of skin sensory nerves [42,62]. This phenomenon likely has both neural and humoral components, but the relative contribution of these to the protection observed is unknown. Most of these non-ischemic cardiac conditioning stimuli are capable of initiating both an immediate protection (i.e., early phase) and a longer lasting protection (i.e., late phase). As will be seen, all of the non-ischemic remote initiators of conditioning seem to involve neural and hormonal mechanistic aspects. In this, they are similar to RIPC. However, remote perconditioning and postconditioning are more similar to the early phase in their timing of action (immediate onset of protection).
《3.1. Spinal cord stimulation》
3.1. Spinal cord stimulation
Activation of the spinal dorsal horn neurons by spinal cord stimulation (SCS) is known to be effective in treating chronic intractable angina pectoris. In addition to pain relief, clinical studies using SCS for the treatment of chronic refractory angina demonstrate increases in exercise tolerance, improvements in ischemia-related EKG changes, and improvements in the quality of life [37,63,64]. Animal studies also indicate that SCS improves the function of the heart. It has been demonstrated that SCS reduces infarct size in a rabbit model with transient coronary artery occlusion, an effect that can be prevented by adrenergic blockers [65]. However, this effect was not observed when performed as a postconditioning stimulus. This result, along with the invasive nature of the procedure, limits its clinical applicability, although SCS is being studied and has other potential uses, such as in heart failure (HF) and arrhythmia.
《3.2. Vagal stimulation》
3.2. Vagal stimulation
It has long been recognized that balance of the autonomic nervous system is critical to many aspects of homeostasis and functional regulation in the heart. After myocardial ischemia, there is an imbalance between the sympathetic and parasympathetic systems, and increased chronic sympathetic activity is thought to contribute to mortality after MI and in HF [66−68]. Vagal stimulation has been studied for its potential to slow left ventricle (LV) remodeling and improve heart function and survival in post-MI HF. Based on this potential, Kong et al. [38] showed that vagal stimulation reduces infarct size and improves function in a rat MI model. The mechanism they describe includes the activation of protein kinase C (PKC), nitric oxide (NO) production, and anti-inflammatory effects that are not dissimilar to those that have been described after IPC and RIPC [38]. It has been demonstrated that transection of the femoral nerve likewise abolishes the cardioprotection of RIPC [61] and that stimulation of the femoral nerve can mimic RIPC of limb ischemia as well [39], supporting the concept that neural pathways are common to many forms of cardiac conditioning and that direct stimulation of some nerves can elicit cardioprotection.
《3.3. Electroacupuncture (EA)》
3.3. Electroacupuncture (EA)
In 2003, Tsou et al. [69] showed that EA at the Neiguan acupoints (corresponding to a part of the human wrist area, innervated by the thoracic vertebral region) reduces I/R injury, measured by reduced cardiac enzymes in a rat model. They also showed that this effect was blocked by the severing of the bilateral median nerve, the bilateral severing of the vagus, or by IV naloxone, implicating neural pathways, including the vagus, whose stimulation is already known to be capable of conditioning. Wang et al. [70] demonstrated that EA induces cardiac conditioning against MI and that this effect adds to the protective effect of hypothermia, suggesting complementary mechanisms. In 2006, Gao et al. [40] showed that EA at bilateral Neiguan acupoints is cardioprotective, reducing infarct size in a rat model by nearly 50%. This effect was blocked by propranolol, revealing dependency upon the adrenergic signaling. In 2012, Zhou et al. [41] showed in a rabbit model that perconditioning by EA (EA during coronary occlusion) reduced infarct size concomitant with the release of cardiac norepinephrine (NE), and that this protection was reduced by the pharmacologic blockade of PKC and opioid receptors in an additive manner. This work implicates many of the same signaling pathways involved in IPC/RIPC, as well as the neural signaling in EA-induced conditioning. Redington’s group, which had previously shown the involvement of a dialyzable circulating factor in RIPC [61], showed that such is also produced in the rabbit after bilateral EA at the Neiguan points [71]. Despite more than 15 years of research, the humoral factor(s) have never been identified. Other work has implicated roles for Hsp70 (also critical for IPC and RIPC [72,73]) and miR-214 (also involved in ischemic postconditioning [74,75]), supporting the idea that in addition to common signaling and neural pathways, gene regulatory pathways may be common to these different forms of cardiac conditioning. To date, EA has not been shown to be effective as a postconditioning stimulus, but the perconditioning protection may have clinical relevance [41].
《3.4. Initiating cardioprotection by sensory nerve activation: RPCT and NIC》
3.4. Initiating cardioprotection by sensory nerve activation: RPCT and NIC
In 2004, we published a paper describing a cardioprotective effect of skin surgical incision and coined the term remote preconditioning of trauma (RPCT) for the effect. Briefly, we found that a surgical incision in the abdominal peri-umbilical region elicited cardioprotection against a subsequent ischemic insult, resulting in an 80% to 85% reduction of cardiac infarct size. Unlike either IPC or RIPC, RPCT is ischemia-independent and yet is highly efficacious. Like RIPC, RPCT requires intact neural connections [42]. RPCT has both an early and late phase, like IPC; however, unlike IPC, both the early and late phases of remote preconditioning of trauma are TNF-α and iNOS independent [42,62,76]. This shows that RPCT is not mechanistically identical to IPC; however, as we will see, there is mechanistic overlap.
An unusual aspect of RPCT is that the initiation of the protective phenomenon is associated with peripheral nerve stimulation of a particular site—the skin bilaterally located around the umbilicus [42,62]. We showed that this stimulus acts though skin sensory nerves, as the effect is blocked by topical lidocaine and mimicked by topical capsaicin cream, with the effect of topical capsaicin being completely blocked in TRPV1 knockout mice [42]. This result implicates neural pathways, and indeed, we showed that transection of the spine at the T7 vertebral level blocks protection, while C7 transection does not, demonstrating a lack of central nervous system (CNS) regulation, but the necessity of spinal integrity between the umbilicus and the heart. Based upon the fact that skin nociception is the initiator of this protection, and that chemical and other pain initiators can activate it, we have coined the term nociception-induced conditioning (NIC) to refer to the broader phenomenon (activated by incision, chemical, or as we shall see, electrical means).
Further work showed that the neural pathway could be traced via injection of a fluorescent dye, 1,1’-dioctadecyl-3,3,3’,3’-tetramethylindocarbocyanine (DiI) at the skin incision site. The DiI signal was shown to stain the dorsal horns at both the T1−T5 (abdomen) and T9−T10 (cardiac) vertebral levels. This result suggests a neural pathway from the skin sensory nerves to the spinal dorsal root ganglia (DRG) at the level of the skin incision to the level of the cardiac innervation [42]. The literature supports the idea that a dorsal root reflex induced by peripheral nociceptive stimulation may form the basis of the interactions between the somatosensory and cardiac sympathetic systems at different spinal levels [77,78]. The dorsal root reflex is a volley of action potentials that can be recorded emerging antidromically from the spinal cord in the dorsal roots following the electrical stimulation of an adjacent dorsal root or sensory nerve. The dorsal root reflex is thought to arise on the rising phase of the primary afferent depolarization (PAD) generated within the terminals of afferent fibers in the cord by a synchronous afferent volley arriving in the cord after electrical stimulation of sensory nerves and roots [79,80]. This causes depolarization of the terminals of the afferent fibers within the cord beyond their threshold, producing action potentials that travel antidromically out of the cord along the dorsal roots. Spreading of the dorsal root reflex both along and across the cord has been noted by several groups [81,82]. In an isolated hamster spinal cord, the dorsal root reflex has been shown to spread over at least fifteen segments from the lower lumbar to upper thoracic roots, in both rostro-caudal and caudo-rostral directions [82]. This suggests the existence of a pathway within the mammalian cord for the transmission of PAD between segments. The involvement of this pathway is supported by our spinal transection results [42]. The fact that both Hoe 140 and propranolol block cardioprotection, as does the genetic ablation of BKR2, supports the involvement of bradykinin and adrenergic signaling, as was found for IPC and RIPC [59,60]. We also showed a requirement for PKC activation and activity of the mitoKATP channel, supporting the hypothesis that common mediators and end-effectors are involved in NIC [42] and IPC [83]. The blockade of calcitonin gene-related peptide (CGRP) and substance P signaling supports a pathway whereby the antidromal activation of cardiac sensory nerves activates cardiac sympathetic nerves. NE and bradykinin likely then act upon cardiomyocytes and are the activation signals for PKC. The Gross’s lab and others [84,85] have confirmed that RPCT (which we now see as one sort of stimulus for NIC) operates in dogs and rats, and have confirmed the requirement of bradykinin signaling. While the pharmacologic blockade of adenosine and opioid signaling had no effect upon cardioprotection, unlike in IPC. Gross et al. showed that signaling by epoxyeicosatrienoic acids (EETs) are important downstream of bradykinin signaling in NIC (induced by either RPCT or topical capsaicin) and have previously shown EETs to be involved in IPC and ischemic postconditioning [85−87]. Although EETs are thought to act locally in an autocrine or paracrine manner, longer-distance actions cannot yet be ruled out in NIC. Finally, using a topical inhibitor of PKCγ [88], which is a neuron-specific PKC isoform, they blocked NIC, confirming the role of skin nerve fibers and providing a new pharmacologic target for inducing NIC [86].
We believe that the published evidence supports a mechanism for NIC whereby the activation of skin nociceptors at the anterior abdominal location (near T9−T10) sends a signal from the DRG at T9−10 to the DRG at T1−5, and that this initiates an antidromic signal via cardiac sensory nerves to the heart. In the heart, the sensory nerves would release CGRP and substance P, which would trigger the release of NE from sympathetic nerves and bradykinin from either the sensory or sympathetic nerves [89]. NE and/or bradykinin would act in part upon cardiomyocytes, triggering PKC activation that would mediate the signaling and activation of end-effectors, including the mitochondrial KATP channels [42,85]. This sympathetic system-mediated cardiac conditioning is triggered in response to “warning signals” relayed by the cord from the remote skin nerves.
Although there is a great deal of overlap between the mechanisms of IPC, RPCT, and NIC, it is notable, based on our limited data at present, that NIC does not employ TNF-α, adenosine, or opioid signaling [62,85], while IPC requires each of these. Interestingly, sympathetic activation, bradykinin, and β-adrenergic signaling have been implicated in NIC and IPC, PKC seems to be required for all of these conditioning modalities, and CGRP has been shown to be cardioprotective, and to be involved in RIPC, postconditioning, and cardioprotection after heat stress [90−93]. Overall, the available evidence supports the hypothesis that cardiac conditioning phenomena share aspects of mechanism while not requiring completely identical molecules. We propose that this is indeed the case and that different cardioprotective phenomena essentially represent different entry points into a large signaling network consisting of receptors, kinase and metabolic signaling cascades, gene networks, and effector pathways (Figure 1). Each type of conditioning signal activates this network at a distinct “edge” of the network, with the consequence that specific portions of it are the focus of detectable signaling pathways that converge upon more common protective effector pathways. The common mediator and effector pathways seem to include bradykinin (discussed above), PKC, and mitoKATP.
《Fig. 1》
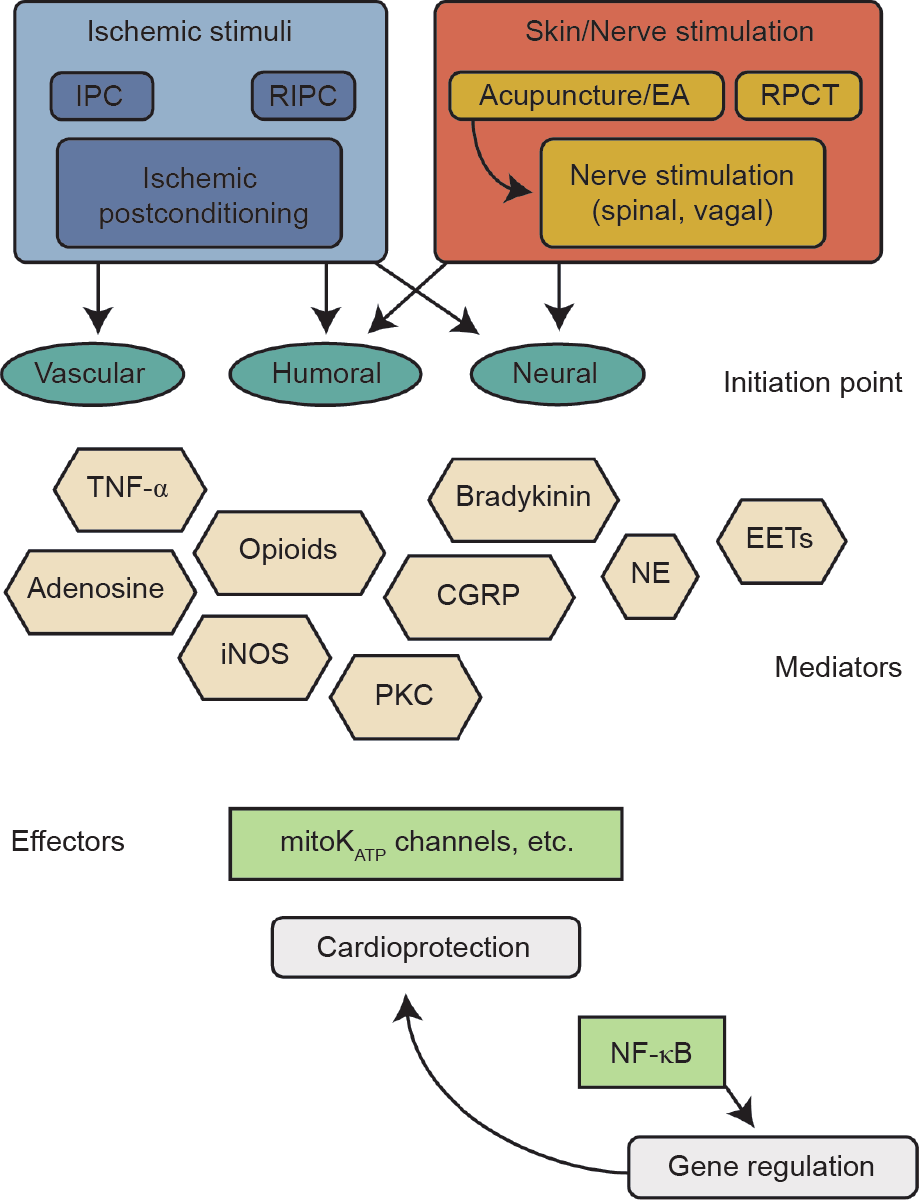
Fig.1 The “Cardiac Conditioning Network” construct postulates that a portion of the cellular informational network is activated on different edges by different stimulus, leading to convergent signaling that results in myocardial salvage. The conceptual diagram is not meant to be comprehensive, but to point out similarities that convey the concept. Abbreviations: IPC, ischemic preconditioning; RIPC, remote ischemic preconditioning; EA, electroacupuncture; RPCT, remote preconditioning of trauma; TNF-α, tumor necrosis factor alpha; iNOS, inducible nitric oxide synthase; CGRP, calcitonin gene-related peptide; PKC, protein kinase C; NE, norepinephrine; EETs, epoxyeicosatrienoic acids; mitoKATP, mitochondrial potassium ATPase; NF-κB, nuclear factor kappa B.
《3.5. Protein kinase C (PKC)》
3.5. Protein kinase C (PKC)
PKC plays an integral role in many of the signaling cascades that govern cellular behavior [94]. The current paradigm of PKC signaling in cardioprotection has been primarily formulated on the basis of information acquired through the pharmacologic inhibition of various signaling elements. In support of this linear paradigm of PKC signaling, the activation of multiple proteins in a PKC-dependent fashion has been demonstrated in response to IPC [95] and RIPC [96], indicating that these proteins may be signaling elements in the PKC pathway. As it is one of the most highly expressed myocardial PKC isoforms [95], PKC-α is the least studied because, unlike the novel isoforms PKC-δ and-ε, it is not regulated in acute myocardial ischemia. Translocation modification has confirmed that PKC-δ is a critical mediator of post-ischemic cardiomyocyte necrosis and contractile dysfunction in mice, rats, and pigs [97,98], although the activity in the porcine model is disputed by one study [99]. In the normal myocardium, PKC-ε forms signaling complexes with at least 36 different proteins that can be classified as structural elements, signaling molecules, and stress-responsive proteins [100]. PKC-ε dependent cardioprotection induces dynamic modulation of these complexes, suggesting a functional role for these complexes in the genesis of protection against ischemic injury [94,101].
《3.6. Mitochondrial/sarcolemmal KATP channels》
3.6. Mitochondrial/sarcolemmal KATP channels
Mitochondria play an important part in myocyte energy production through oxidative phosphorylation. They are also involved in key aspects of cardiomyocyte survival, through regulating intracellular reactive oxygen species (ROS) generation, calcium homeostasis, and the release of apoptosis-inducing factors [102−104]. Both mitoKATP and sarcKATP channels are present in the mammalian heart [105] and appear to be involved in IPC and cardioprotection [102]. Gaudette et al. [106] demonstrated in a sheep model that protection provided by direct mitoKATP openers could be abolished by PKC antagonists (chelerythrine); and that protection mediated via the activation of PKC could be abrogated by administration of the mitoKATP channel antagonist 5-hydroxydecanoate (5-HD), a selective blocker of the mitoKATP channels, implying that the activation of PKC and of mitoKATP channels are both codependent and necessary for protection in the heart. Nozawa et al. [107] showed that IPC in rats produced a translocation of both PKC-ε and PKC-δ; however, 5-HD blocked the effect of IPC without blocking the translocation of the two PKC isoforms. These results suggest that the mitoKATP channel or the site of action of 5-HD is distal to PKC. The mitoKATP is one of the major and likely common effector pathways for improving cell survival after I/R injury. However, as for other pharmacologic inducers of cardiac conditioning, severe limitations exist on the clinical utility of drugs targeting this channel.
《4. The potential clinical utility of NIC and EA》
4. The potential clinical utility of NIC and EA
As mentioned earlier in this paper, EA has not been shown to be useful for cardiac postconditioning; however, one positive study supports its use as a perconditioning stimulus [41]. Because of mechanistic similarities, and because of the limited clinical utility of RPCT (i.e., involving surgical skin incision), we sought an easier, non-traumatic way to initiate NIC by abdominal sensory nerve activation using electrical stimulation—EA of the skin in this instance. We tested the hypothesis that remote electrical stimulation of the same skin field stimulated using RPCT and NIC (with capsaicin [42]) in our previous work could be used to activate cardiac conditioning, and herein report our results, for the first time.
We performed electrical skin stimulation (ES) using electroacupuncture needles and current (for technical details, see the legend of Figure 2) 15 min prior to I/R or at the beginning of reperfusion (for 15 min thereafter). For ES, an EA needle was not inserted deep into the tissue as it is for acupuncture, but was inserted and only hooked through the skin (thus, the term ES). Infarct size was used to evaluate the results of I/R injury with or without prior ES treatments. The results of this study demonstrate that ES of specific points spaced equally along the line lateral to the umbilicus (the same region used for RPCT) induces a powerful 80%−85% ( P≤0.05) reduction in infarct size when applied either as a preconditioning or as a postconditioning stimulus (Figure 2). This result supports the conclusion that NIC by ES at the anterior abdominal site is efficacious against I/R injury in the mouse, similar to RPCT. Since this intervention works as a postconditioning stimulus, is non-traumatic, non-ischemic, and does not require surgical intervention, we believe it has great clinical relevance.
《Fig. 2》
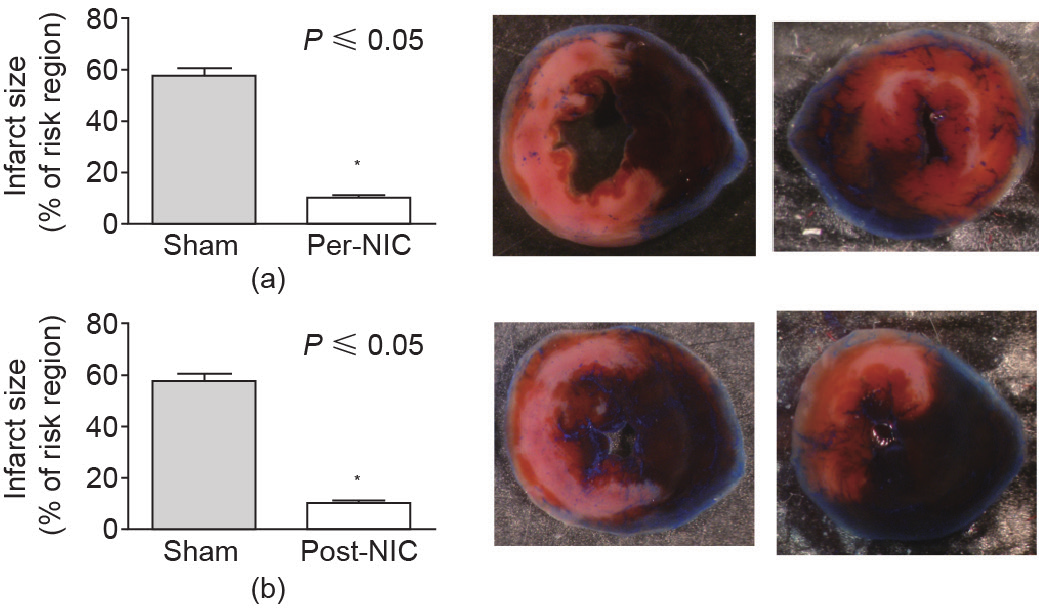
Fig.2 Cardiac conditioning by nociceptor activation via electrical skin stimulation (ES). Sterile finger and facial acupuncture needles (0.18 × 7 mm; Millenia Acupuncture, Alhambra, CA, USA) were placed equally on a line through the region previously delineated for RPCT [62], connected to a KWD-808-I multi-purpose health device (Yingdi Inc., China) and stimulated with electricity (10 V 100 Hz pulse width 400 ms) for 20 min for preconditioning, and then ischemia was initiated ((a) Pre-NIC). For (b) post-NIC, the stimulus was initiated just prior to reperfusion and continued for 20 min. Needles were removed before the mice regained consciousness and reperfusion was for 24 h. At the end of reperfusion, the mice were anesthetized and the hearts were cannulated and stained, fixed, photographed, and the infarcts analyzed, as previously detailed [42,62]. Right: Representative TTC stained mid-LV images from sham (top and bottom left), pre-NIC (top right), and post-NIC (bottom right). Reduction of infarct size was greater than 80% for both pre- and post-NIC.
《5. Conclusions》
5. Conclusions
Nearly three decades of studies have taught us much about the mechanisms that underlie the endogenous cardioprotection afforded by IPC. The discovery of RPCT has offered significant therapeutic strategies for protecting the heart against myocardial I/R injury, and opened the door to the exploration of new modalities for triggering cardioprotection. The fact that some of these modalities work in multiple model species, and in humans [108], supports that these models are worthwhile laboratories to further our understanding and for preclinical development. Mechanistic and combinatorial studies support the hypothesis that preconditioning, perconditioning and postconditioning, as well as IPC, RIPC, nerve stimulation, EA, and NIC have many common mechanistic components. Yet mechanistic differences remain as well. For example, both IPC and RIPC involve the action of TNF-α, adenosine, and opioids, and yet these are not involved in the mechanism underlying RPCT [62] and NIC [85]. EA has been reported to be effective as a preconditioning but not as a postconditioning stimulus [41]. Yet all of these modalities require and employ the activity of NF-κB, PKC, bradykinin, and the mitoKATP channel [42,62]. We propose that all of these cardioprotective phenomena are in fact a reflection of a broader cardiac conditioning network, and that each stimulus works through slightly different initiating points in that network (Figure 1). IPC seems to invoke multiple initiating points, including vascular, humoral, and neural, while NIC and EA work primarily through the neural initiating portions of the network. Differences in initiation may relate to differential activation of components of the network; thus, their detectable activation, necessity, or dispensability in the particular phenomena under study are different. Our view of these phenomena as discrete derives primarily from the timing over the last 29 years of our experimental discovery of the different modalities and the mechanistic pieces. The proposed conditioning network includes: humoral factors; neural and vascular signaling; kinase, oxidative, and metabolic cascades in cardiac cells (and perhaps between cells of different types); gene expression; and related miRNA networks. As far as we can tell, these network components converge at several points, including PKC, bradykinin, and the mitochondria. As our work progresses, we predict that we will find additional components of the network that are more critical for NIC, ES, and RIPC, for example, but that are dispensable for IPC. We recognize that the existence of such a conditioning network is really a human construct that designates parts of the overall cellular and intercellular informational network as having a specific function related to myocardial salvage. However, for the purposes of research and translation, such constructs are helpful to delineate functions for molecules that may eventually be considered as therapeutic targets.
There remains a great clinical need for non-ischemic and non-traumatic methods to initiate cardiac conditioning. At the moment, the discovery of NIC and EA/ES-induced neural pathways as remote non-ischemic initiators holds great promise for the development of pharmacologic and electroceutical approaches [109,110] to elicit cardioprotection without causing ischemia or requiring surgical intervention. We demonstrate herein for the first time that NIC is protective as a postconditioning stimulus, establishing the clinical relevance of studying the underlying mechanism and further preclinical development.
《Acknowledgements》
Acknowledgements
This study was financially supported by grants from the National Institutes of Health (NIHR01 HL091478) for W. Keith Jones and the National Natural Science Foundation of China (81470425) for Xiaoping Ren.
《Compliance with ethics guidelines》
Compliance with ethics guidelines
CardioCeption, LLC holds licenses relevant to this field of study. W. Keith Jones owns equity in CardioCeption, but has received no income or financial benefit from any entity related to this work. The University of Cincinnati, W. Keith Jones, and Xiaoping Ren filed the initial patents but have received no royalties.
Kristin Luther, Yang Song, Yang Wang, Xiaoping Ren, and W. Keith Jones declare that they have no conflict of interest or financial conflicts to disclose.











 京公网安备 11010502051620号
京公网安备 11010502051620号




