
《1.Introduction》
1.Introduction
Trillions of gut microbes reside in the human body, primarily within the gastrointestinal tract (GIT), where the population of gut microbes increases by approximately eight orders of magnitude from the proximal small intestine (103 mL–1 luminal content) to the colon (1011 g–1 content) [1]. Taking a 70 kg man as a reference, the total number of microbes is estimated to be 3.8 × 1013, with a total weight of 0.2 kg [2]. Metagenomics sequencing is able to detect more than 1000 species of gut microbes, which are dominated by four major phyla: Firmicutes, Bacteroidetes, Actinobacteria, and Proteobacteria. The genomes of these microbes encompass more than three million genes—approximately 100 times more than the entire human genome [3]. These gut microbes are acquired as early as during fetal development, and there is growing evidence for the presence of gut microbes in the placenta, amniotic fluid, and meconium [4–7]. Although gut microbes are phylogenetically conserved [8], the composition of human gut microbes can be affected by the host’s personal hygiene, diet, drug intake, and disease status throughout his or her life [9,10]. These effects are more profound during the early life stages, when initial microbial colonization begins; however, the stability and homeostasis of gut microbes are usually established once the host is between 2–5 years of age [11].
In recent years, gut microbes have been gaining increasing attention due to their impact on human health [12]. The composition and function of our gut microbes appear to play an essential role in almost every biological process in the body. It has long been recognized that gut microbes can help their host to defend against pathogens, develop a healthy intestinal structure and immune system, and aid with the digestion of indigestible dietary fibers [13]. More recently, associations have also been recognized between gut microbes and diseases such as fatty liver disease, coronary heart disease, cancer, and obesity and diabetes [14]. In this review, we summarize and provide the most recent facts on the roles of gut microbes in human health, along with the unmet need for and solutions toward modulating the gut microbiota.
《2.The function of gut microbes in human health》
2.The function of gut microbes in human health
Gut microbes can coexist symbiotically with a healthy human. Short-chain fatty acids (SCFAs), including butyrate, acetate, and propionate, are fermentation products of dietary fibers produced by gut microbes. Although such fibers are otherwise indigestible by humans, their metabolites provide essential nutrients for colonic cells and play an important role in maintaining gut health. Eubacterium hallii (E. hallii) is considered to be an SCFA-producing microbe, and a recent discovery indicates that E. hallii is capable of producing high levels of glycerol/diol dehydratase, a key enzyme in the conversion of glycerol to 3-hydroxypropionaldehyde, a process that also produces cobalamin [15]. Levels of serotonin, a neurotransmitter synthesized in colonic enterochromaffin cells that is known to regulate a wide range of physiological activities such as enteric motor and platelet function, are regulated by the ingestion of spore-forming bacteria, mainly consisting of clostridial species [16]. Other studies have also investigated the relationship between gut microbes and gut metabolites.
Gut microbes are thought to metabolize phenyl-containing amino acids, such as tryptophan. Levels of tryptophan metabolites, such as indoxyl sulfate and indole-3-propionic acid, are highly correlated to the presence of gut microbes [17]. The production of indole-3-propionic acid is dependent on the colonization of the bacterium Clostridium sporogenes [17]. Furthermore, variations in Faecalibacterium prausnitzii colonies have been associated with levels of urinary metabolites involved in multiple metabolic pathways [18]. One such association has been identified between Clostridium difficile (C. difficile) and levels of fecal cholesterol and coprostanol, indicating that gut microbes play a crucial role in lipid metabolism [19]. Taken together, these studies highlight the essential role of gut microbes in maintaining normal physiology in humans. A deviation from the normal gut ecosystem could therefore be associated with a range of abnormalities, such as cancer, nonalcoholic fatty liver disease (NAFLD), obesity and diabetes, coronary heart disease, kidney dysfunction, and neurodegenerative diseases (Fig. 1); these associations will be summarized in the forthcoming sections.
《Fig. 1》
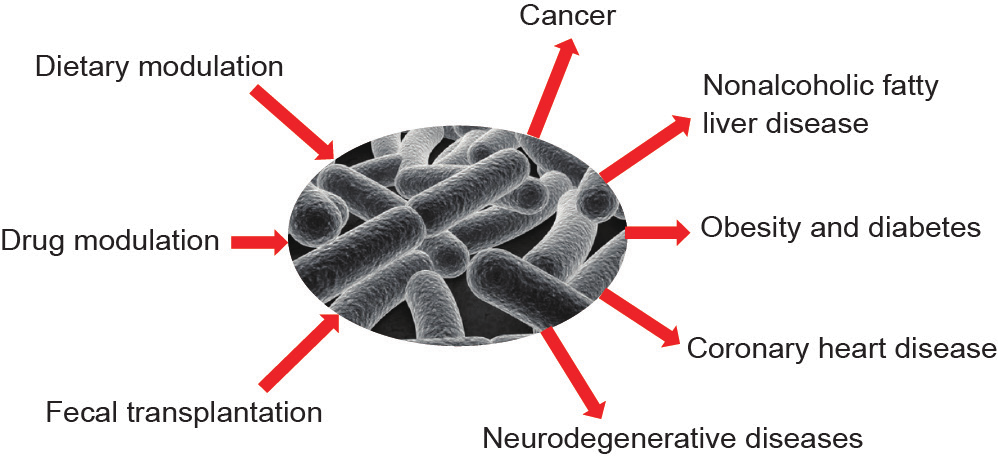
Fig.1. Summary of the role of gut microbes and their modulation in a pathological state.
《2.1. Cancer》
2.1. Cancer
Levels of Helicobacter pylori are widely associated with an increased risk of gastric [20] and colorectal cancer [21]. A Gramnegative bacterium resides in the human stomach and can cause various diseases including gastritis, peptic ulcer, and gastric cancer [21]. Other resident gut microbes have also been identified as major factors in the increased risk of human colon cancer, including Streptococcus bovis [22,23], Bacteroides fragilis [24], and Escherichia coli (E. coli) [25]. These bacteria can cause inflammation and may trigger inflammatory bowel disease, thereby promoting the development of colorectal cancer. The proposed mechanism for this increased risk is recognized as involving Toll-like receptors (TLRs) and triggering downstream signaling pathways such as NF-κB, ERK, JNK, and p38, leading to an up-regulation of the growth factors and inflammatory mediators that promote neoplasia [26].
《2.2. Nonalcoholic fatty liver disease》
2.2. Nonalcoholic fatty liver disease
NAFLD is the most common chronic liver condition in both Western [27] and Asian [28] countries, and is considered to be a hepatic manifestation of metabolic syndrome. Due to its close link with obesity, the pathogenesis of NAFLD has been widely accepted to be the result of multiple “hits,” including the contribution of the intestinal dysbiosis that is associated with obesity [29,30].
Studies show that a shift in the gut microbe composition is associated with obesity, and correlates closely with the prevalence and progress of NAFLD [31–34]. Zhu et al. [34] have suggested that the connection between gut-derived endogenous alcoholic and nonalcoholic steatohepatitis (NASH) might be involved in the pathogenesis of obesity in children. A significant compositional shift in gut microbes, such as the decrease of some selected members of Firmicutes, has been observed in patients with NAFLD [33]. A decrease in Bacteroidetes has also been found among obese patients with NASH compared with healthy controls [34]. In addition, Wang et al. [30] assessed the fecal microbial composition and its correlation with liver biochemistry in non-obese adult patients with NAFLD. Boursier et al. [35] have suggested that the severity of NAFLD is associated with gut dysbiosis along with a shift in metabolic function of the gut microbiota. Boursier et al. [35] also revealed that Bacteroides and Ruminococcus are independently associated with NASH and significant fibrosis, respectively. These studies indicate that an alteration in the composition of the gut microbiota is closely associated with the development of NAFLD.
Several different mechanisms have been uncovered that relate to gut microbe composition and the pathogenesis of NAFLD. Firstly, gut microbes have the potential to increase energy extraction from ingested food [36]; alter appetite signaling [37,38]; and enhance the expression of genes involved in de novo lipogenesis, β-oxidation, or inflammation-driven liver steatosis [39,40]. Secondly, gut microbes and the translocation of gut-derived bacteria or metabolites are likely to influence the development of hepatic inflammation [41,42]. Small intestinal bacterial overgrowth syndrome (SIBOS) is highly prevalent in patients with NAFLD [43], which appears to be related to an increased gut permeability in these patients [44]. Intestinal permeability plays an important role in the pathogenesis of NAFLD, which in turn leads to hepatocellular inflammation [44,45] and insulin resistance [32,39]. It is notable that the expression levels of the lipopolysaccharide (LPS) receptor, CD14, and of TLR-4 are also higher in NASH patients with SIBOS [46]. This finding suggests that SIBOS, and particularly its Gram-negative bacteria population, promotes the progression of NASH through TLR-4. Research has indicated that an up-regulation of CD14 in Kupffer cells is related to a hepatic hyper-inflammatory response to LPS during NASH progression [47].
《2.3. Obesity and diabetes》
2.3. Obesity and diabetes
A large body of evidence from both population and animal studies indicates that alterations in the gut microbe composition are associated with obesity and type II diabetes (T2D). For example, Bäckhed et al. [48] showed that wild-type mice gained 40% more body weight than germ-free mice, although germ-free mice consumed 29% more food, while re-conventionalized germfree mice gained 57% in body weight. This observation led to increased interest in investigating the composition of gut microbes in obese mice. Studies have shown that microbe composition in obese mice (ob/ob) is reduced by 50% in the presence of high levels of Bacteroidetes and Firmicutes, as compared with that in corresponding lean mice [49]. Further evidence revealed that dietinduced obesity may produce rich levels of Firmicutes, specifically in the Mollicutes class. In addition, lean germ-free mice receiving gut microbes from diet-induced obese mice have more fat depositions than those receiving from lean donors [50]. The effect of lean-like gut microbe transplantation is equally significant for obese mice when they are co-housed with lean mice, with the composition of the gut microbes of obese mice containing more Bacteroidetes when such mice are co-housed with lean mice [51].
Population studies have also highlighted the association between gut microbe composition and obesity. Turnbaugh et al. [50] showed that human obesity was associated with alterations in levels of Bacteroidetes and Firmicutes. It is notable that obesity-associated microbes from humans can be transferred to germ-free mice, resulting in significantly increased body fat as compared with germ-free mice colonized with lean microbes [36]. The transferable colonization of fator lean-associated microbes has been confirmed by Zhang and coworkers [52]. Obese children and/or children predisposed to obesity, such as those with Prader-Willi syndrome, are subjected to a diet rich in non-digestible carbohydrates in order to induce weight loss [53]. Germ-free mice colonized with pre-intervention gut microbes showed larger body fat accumulation as compared with mice with post-intervention gut microbes [52]. Alterations of gut microbe composition are also associated with T2D [54]. An overall decrease in proportions of phylum Firmicutes and class Clostridia is presented in diabetic patients, which is in contrast to gut microbes found in obese patients [55]. In addition, Clostridium species and Lactobacillus species appear to be important in driving T2D [56]. The range of SCFA-producing bacteria is low in the diabetic population as compared with a healthy background population [3].
The link between gut microbe composition and obesity is believed to be mediated by the metabolic function of the gut microbes. That is, gut microbes produce SCFAs, which provide essential nutrients and energy for maintaining a healthy and intact gut barrier. Without these gut microbes, the GIT becomes defective, possibly due to the lack of essential SCFAs. Such a defective gut structure may inhibit the delivery of nutrients to the liver, stopping them from being converted into fat for deposition. Another important function of gut microbes is the modulation of lipid absorption. This process is achieved via the action of gut microbes that are capable of removing polar groups of glycine and taurine from bile acids, leading to less fat being emulsified and absorbed from the GIT. De-conjugated bile acids are therefore absorbed by the host, affecting cholesterol metabolism [57,58]. Kishino et al. [59] identified genes in the gut bacterium Lactobacillus plantarum that encode for the enzymes involved in the saturation metabolism of polyunsaturated fatty acids.
《2.4. Coronary heart disease》
2.4. Coronary heart disease
Gut microbes associated with obesity and T2D may promote coronary heart disease. In addition, bacterial DNA is found in human atherosclerotic samples; to be specific, Chryseomonas is present in human atherosclerotic samples, and Veillonella and Streptococcus are found in the majority of atherosclerotic samples, as well as in the oral cavity and gut of the same patients [60]. This evidence indicates that the presence of these gut microbes correlates directly with coronary heart disease. Another population study involving 893 participants constructed a novel model demonstrating that 26% of high-density lipoproteins are attributed to the gut microbiome, independent of age, sex, and host genetics [61]. Gut microbes modulating lipid metabolism and cholesterol via bile acids provide a possible mechanism for the relationship between gut microbes and coronary heart disease. A link between coronary heart disease and the microbial metabolism of dietary choline and L-carnitine has already been established [62,63]. Both choline and L-carnitine can be metabolized by gut microbes, producing trimethylamine (TMA), which is then further metabolized in the liver into trimethylamine-N-oxide (TMAO). TMAO has been recognized as a pro-atherogenic agent [64]. The underlying mechanism that describes the association between TMAO-producing gut microbes and coronary heart disease is attributed to the action of a TMAO-producing enzyme, flavin monooxygenase-3, which is involved in the perturbations of reverse cholesterol transport and which has the effect of reorganizing the total cholesterol balance of the body [65,66]. Further evidence has shown that targeting the gut microbial production of TMA with a non-lethal microbial inhibitor, 3,3-dimethyl-1butanol, could reduce the levels of TMAO and prevent atherosclerotic lesion [67].
《2.5. Neurodegenerative diseases》
2.5. Neurodegenerative diseases
Increasing evidence is revealing the involvement of gut microbes in modulating neurological diseases via a bidirectional microbiota-gut-brain axis [68]. In experiments, germ-free mice exhibited increased motor activity and reduced anxiety-like behavior compared with conventional specific pathogen-free mice [69]. The germ-free mice also expressed lower mRNA levels of hippocampal serotonin 1A receptor and amygdala N-methyl-Daspartate receptor (NMDAR) subunit NR2B [70], and higher levels of the brain-derived neurotrophic factor (BDNF) in the dentate granule layer of the hippocampus [70]. Other studies have found that colonization with Clostridium butyricum can restore cognitive defects [71]. Administration of Lactobacillus farciminis [72], Lactobacillus reuteri [73], Bacteroides fragilis [74], or Lactobacillus rhamnosus strains [75,76] could also potentially prevent stressinduced social deficit and reduce the levels of stress-induced plasma corticosterone in mice [76], suggesting that the gut microflora could be a potential therapeutic target for neurodegenerative diseases. This result was verified by Möhle et al. [77], who used a broad spectrum of antibiotics (such as ampicillin and vancomycin) to treat adult mice with neurodegenerative diseases. Their study concluded that antibiotic treatment decreased hippocampal neurogenesis and memory retention from neuronal progenitors, and that recognition tests were affected. These deficits were, however, fully restored in mice fed with a mixture of eight probiotics, namely Streptococcus thermophilus, Bifidobacterium breve, Bifidobacterium longum, Bifidobacterium infantis, Lactobacillus acidophilus, Lactobacillus plantarum, Lactobacillus paracasei, and Lactobacillus delbrueckii subsp. bulgaricus [77]. It is notable that all of these probiotic modulations were similar in vagotomized mice, demonstrating that the vagus is a major modulatory constitutive communication pathway in connecting bacteria, gut, and brain [76].
Neuroinflammation is likely to be closely linked with multiple neurodegenerative pathways involved in neurodegenerative diseases [78]. The compositions of fecal microbiota from patients with Parkinson’s disease exhibited decreased “anti-inflammatory” butyrate-producing bacteria, such as Blautia, Coprococcus, and Roseburia, whereas the mucosal microbiota displayed reduced levels of Faecalibacterium [79]. Such colonic dysbiosis with a lower abundance of fecal E. coli and Butyrivibrio fibrisolvens was also reported in an amyotrophic lateral sclerosis mouse model [80], implying that bacteria may play a crucial role in the neuroinflammation of neurodegenerative diseases. More evidence for the presence of a gut-brain axis comes from studies on the modulation of gut microbes on neurotransmitters. Barrett et al. [81] revealed that microaerophilic Lactobacillus and Bifidobacterium species were capable of metabolizing glutamate into gamma amino butyric acid (GABA). Furthermore, Cyanobacteria have been reported to produce neurotoxins such as β-N-methylamino-L-alanine (BMAA) that contribute to various neurological dysfunctions [82].
《3.Modulation of gut microbes》
3.Modulation of gut microbes
《3.1. Dietary modulation of gut microbe composition》
3.1. Dietary modulation of gut microbe composition
Gut microbes are increasingly being recognized as a forgotten “organ” that plays a crucial role in disease development. Questions remain as to how microbes can be manipulated in order to maintain good health. Food intake appears to be the first treatment of choice in modulating gut microbes. Amato et al. [83] showed that a Western diet may result in increased levels of Firmicutes and reduced levels of Prevotella, which are both associated with obesity and T2D. In animal models, levels of Firmicutes and Bacteroidetes are closely associated with a high-fat diet [84]. Four weeks of continuous intake of a high-fat diet has been shown to induce an increased abundance of Firmicutes and a decreased abundance of Tenericutes. Reduced levels of Bacteroidetes occur after eight weeks of continuous intake of a high-fat diet [84]. Modification of the resulting unhealthy gut, using a vegetarian diet or probiotics, may be achieved by providing nutrients that are favored by some bacteria groups, such as Prevotella [85]. Interventions with four days of a meat-based or vegetable-based diet have been shown to change the composition of gut microbes extensively. For example, an increased abundance of bacteria involved in amino acid fermentation, such as Alistipes putredinis, Bilophila, and Bacteroides, is associated with a meat-based diet, whereas bacteria involved in carbohydrate fermentation, such as Roseburia, Eubacterium rectale, Ruminococcus bromii, and Faecalibacterium prausnitzii, are associated with a vegetable-based diet [85]. Recent studies into the correlations between gut microbes and 126 exogenous and intrinsic host factors, such as disease states and dietary factors, have identified a positive correlation between a high-carbohydrate diet and high levels of Bifidobacteria, but a negative correlation between a high-carbohydrate diet and Lactobacillus, Streptococcus, and Roseburia species [86]. Furthermore, red wine consumption is associated with increased levels of Faecalibacterium prausnitzii, which is known to have an anti-inflammatory effect [87].
Prebiotics, such as dietary fibers and some oligosaccharides, have been shown to have beneficial effects on human health [88]. Changes in gut microbes as sociated with ingestion of prebiotics have been widely reported. For example, a 10-fold increase in fecal Bifidobacteria has been demonstrated in people who have received oligosaccharides intervention as compared with a placebo control group [89,90]. Furthermore, probiotics supplementation appears to be more effective in infants than in adults [91], a result that is largely due to the gut microbial community, which may have a greater impact on infants since it is under development at that age, resulting in a more easily manipulated effect. Thus, an increased abundance of Lactobacillaceae and Bifidobacteriaceae species has been found in children supplemented with Lactobacillus rhamnosus GG (LGG) [92]. In addition, LGG supplementation induces increased levels of Lactococcus and Lactobacillus, along with a decrease in E. coli in pre-school children [93].
《3.2. Drugs modulate gut microbe composition》
3.2. Drugs modulate gut microbe composition
Antibiotic treatment is used to treat pathogenic infections, and is shown to have profound effects on the gut microbiota. Zhao et al. [94] reported that treating mice with gentamicin and ceftriaxone induces decreased levels of Barnesiella, Prevotella, and Alistipes, albeit with increased levels of Bacteroides, Enterococcus, Erysipelotrichaceae incertae sedis, and Mycoplasma. In addition, mouse models have shown that this treatment can cause a decrease in a range of metabolites, including SCFAs, amino acids, and primary bile acid, and an increase in oligosaccharides, choline, and secondary bile acids [94].
Responses to antibiotic treatment vary between individuals. Analyses of gut microbes over a 10-month period of three individuals who received two courses of ciprofloxacin suggest that the gut microbial community is relatively stable and has limited day-to-day variability, and that large inter-person variations are prominent in the gut microbial community [95]. Moreover, antibiotic treatment results in a rapid shift of the gut microbial community during the course of the treatment, mainly manifested as a loss of microbial diversity. Despite the recovery in the levels of major microbial colonies after a week of antibiotic withdrawal, these levels are not fully restored [95]. This finding reveals that the gut community is resistant to changes unless it experiences repeated perturbations in the long term. Many herbal medicines are rich in polyphenols, which must interact with gut microbes in order to become bioavailable and bioactive [96], and which could modulate the gut microbial community. In addition, herbal medicines often exert mild anti-bacterial effects [97] and, if used over a long-term period, may permanently modulate the gut microbial community. Thus, there is a huge potential for managing diseases through the modulation of gut microbes.
《3.3. Fecal transplantation modulates gut microbe composition》
3.3. Fecal transplantation modulates gut microbe composition
The first description of a fecal microbiota-transplantation (FMT) in the treatment of C. difficile occurred in 1958 [98]. There has been a recent increase in the use of fecal transplantation for modulating the gut microbial community and eliminating C. difficile colonies. Fecal transplantation treatment for C. difficile appears to be effective: 81% of C. difficile infection cases were resolved after the first FMT treatment [99]. This result has been reproduced in a clinical trial of 232 Canadian patients infected with C. difficile, showing that stool preparation methods (freshly prepared vs. frozen) actually had no effect on the outcome of the significance in altering the gut microbial community [100]. This observation demonstrates the benefit of a frozen sample preparation, in which safety measures could be put in place prior to the treatment of patients. The gut microbial communities of donors and their respective recipients are closely correlated soon after the transplantation, but may deviate from each other over time. Thus, populations of Bacteroidetes and Firmicutes in FMT recipients have been shown to increase to levels that are equivalent to those of fecal donors, whereas the levels of C. difficile have been shown to decrease from 4% to 0.2% in FMT recipients [99]. Other microbial alterations have also been associated with fecal transplantation, including the shift from Proteobacteria to Firmicutes and Bacteroidetes dominant species [101–103]. Given the stability of the gut microbial community [95], it may be important for patients infected with C. difficile to receive multiple fecal transplantations in order to establish a healthy microbial system over the long term.
《4.Future perspectives》
4.Future perspectives
This review covered some typical studies on the association between gut microbes and various disease states as well as methods to modulate the composition of gut microbes. It is not our intention to be comprehensive, but rather to highlight some recent and outstanding research publications. At this point, it is evident that gut microbes have a considerable impact on human health. Research has revealed that the gut microbial community may be modulated by various methods, such as food, drugs, and fecal transplantation. It has accordingly become necessary to define the microbial composition of a healthy gut, and to learn how individual characteristics (age, gender, and ethnicity) may affect the composition of the gut microbiota. Given the intrinsic stability of our gut microbial community, long-term interventional follow-up may become necessary as well.
However, more insight is needed into the mechanisms of the action of gut microbes, which are often mediated by metabolites such as bile acids, SCFAs, and choline. The specific microbes responsible for such actions still remain unclear, but are necessary for specific gut microbial modulation. Despite extensive research into correlations between levels of metabolites and specific gut microbes [18], the causative factor of gut microbes in various diseases is not yet clarified. Therefore, investigating these underlying molecular mechanisms remains as an important focus in future research, and will require concerted efforts from scholars in multidisciplinary areas of research, including clinical doctors, microbiologists, molecular biologists, and biological chemists. Finally, novel emerging research on the role of gut microbes in neurodegenerative diseases presents an area of especially unmet need; particular attention is required on how gut microbes and their metabolites can access the brain and affect various neurological functions in humans.
《Acknowledgements》
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (21375144 and 21105115) and the National Basic Research Program of China (2012CB934004).
《Compliance with ethics guidelines》
Compliance with ethics guidelines
Yulan Wang, Baohong Wang, Junfang Wu, Xiangyang Jiang, Huiru Tang, and Ole H. Nielsen declare that they have no conflict of interest or financial conflicts to disclose.











 京公网安备 11010502051620号
京公网安备 11010502051620号




