

《1. Introduction》
1. Introduction
Many of the challenges in the 21st century, from personalized healthcare to energy generation and storage, share a common theme: Materials are at the core of the solution. Traditionally, materials have been discovered by chance or through an empirical process. A typical example is vulcanized rubber, which was prepared in the 19th century based on the observation (from random mixing of compounds) that heating with additives such as sulfur can improve the durability of the rubber. With the great development of first-principles computational methods and tools, as well as the exponential increase of computer power, scientists and engineers can now realistically simulate the properties and behaviors of materials in specific applications and thereby avoid lengthy cycles of formulation, synthesis, and testing. This field—known as computational materials science—is one of the fastest growing areas within the fields of chemistry and materials science. However, although enormous progress has been made in theoretical methods and modeling tools, the size of the theoretical space of all possible chemicals or materials is overwhelming. For example, the number of pharmacologically relevant molecules is estimated to be on the order of 1060 [1]. Therefore, it is impossible to find a strategy to explore this vast structural space.
With the increase of experimental and computational data, the field of materials informatics (MI) has grown quickly in recent years [2]. One important task of MI is to use existing materials data to predict properties for new materials by employing mathematics and information science methods [3]. The key for achieving this is to build a descriptor model that can predict the property of interest based on a known set of input material-specific features. The quantitative structure–property relationship (QSPR) model is an important descriptor model where the input variables are material structural features. Complex relationships usually exist between the inputs and the output of material properties, which are difficult to handle using traditional linear and nonlinear correlation methods. Thanks to the development of machine learning (ML) methods [4], these complex relationships can now be efficiently modeled.
ML is a branch of artificial intelligence (AI) that aims to build models trained from past data and situations. It has started to play a significant role in materials science due to its ability to learn behaviors and trends from available data without knowing the underlying physical mechanisms. An established ML model can, in turn, be used for materials discovery and design. Some examples of successful applications of ML techniques for materials research include the prediction of steel fatigue strength [5], physical and mechanical properties of alloys [6], electronic bandgaps of perovskite materials [7], catalytic activities [8], and acid dissociation constants [9], as well as the identification of promising porous materials [10], polymer dielectrics [11], mixed oxide catalysts [12], organic light-emitting diode (OLED) materials [13], superconductors [14], and photovoltaic materials [15].
A literature search, depicted in Fig. 1 [16], demonstrates that ML is a rapidly growing area with an increasing number of applications in materials research.
《Fig. 1》
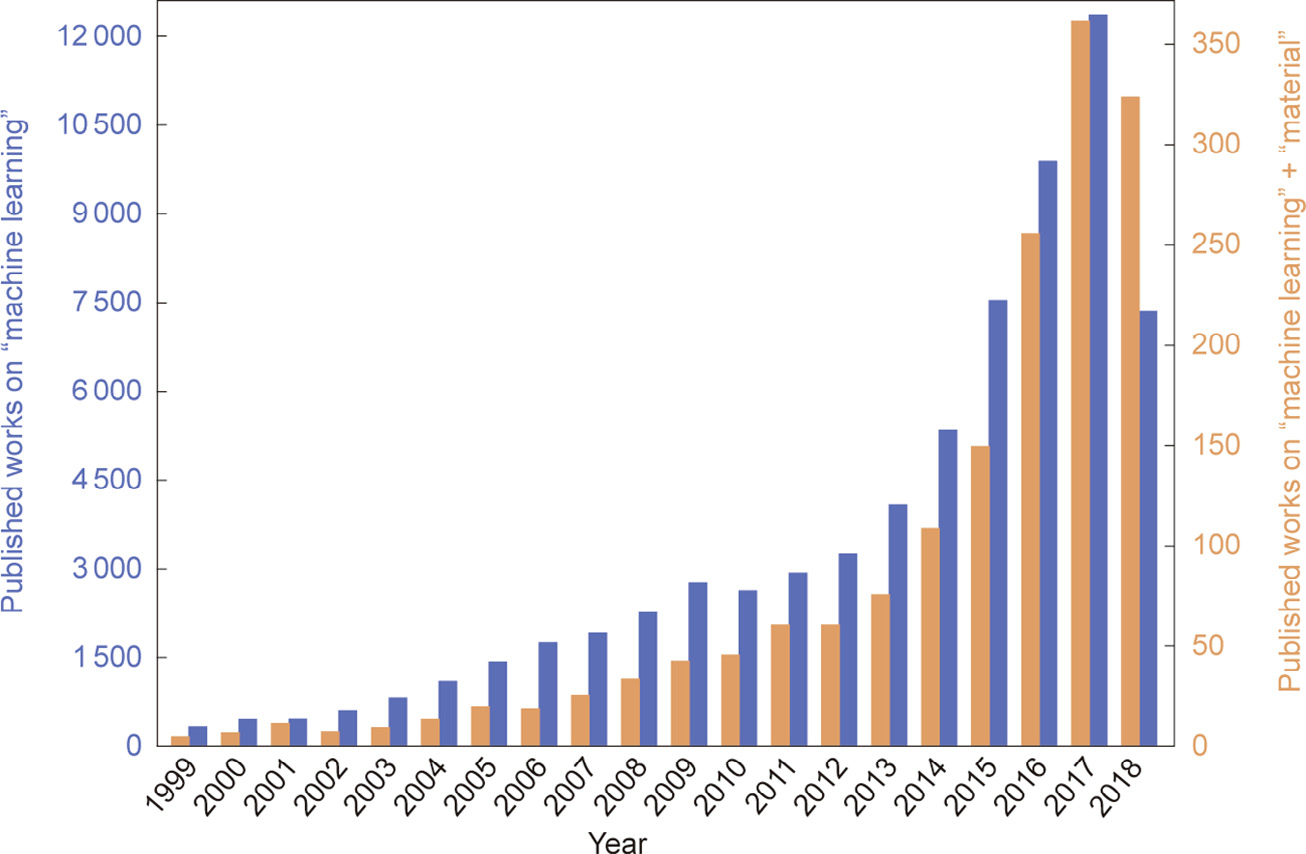
Fig. 1. Number of published works on “machine learning” and “machine learning” + “material” (from January 1999 to September 2018).
Given the increasing importance of data-driven or ML methods in materials research, it is the goal of this review to highlight the main ideas and basic procedures for employing ML approaches for materials research, and to provide an overview on recent important applications of ML for materials discovery and design.
《2. Big data in materials science》
2. Big data in materials science
As illustrated in Fig. 2 [17], for thousands of years, science consisted of empirical observations of natural phenomena. A few centuries ago, the paradigm of theoretical science then arose, characterized by the formulation of various classical laws, theories, and models. With the invention of computers a few decades ago, a third paradigm of science—namely, computational science— emerged, which allows for the simulation of complex real-world problems based on the theories summarized in the second paradigm. Representative examples in materials science are density functional theory (DFT) and molecular dynamics (MD) simulations. The large amount of data generated by experiments and simulations has given rise to the fourth paradigm of science over the last few years: (big) data-driven science, along with the popularization of AI methods. The most important subfield of AI that has evolved rapidly in recent years is ML.
《Fig. 2》

Fig. 2. The four paradigms of science: empirical, theoretical, computational, and data-driven. ANN: artificial neural network; SVM: support vector machine.
There has been an absolute explosion in the amount of published works on “big data” and “data-driven,” as depicted in Fig. 3 [16].
《Fig. 3》

Fig. 3. Number of published works on “big data” and “data-driven” methods (from January 1999 to September 2018).
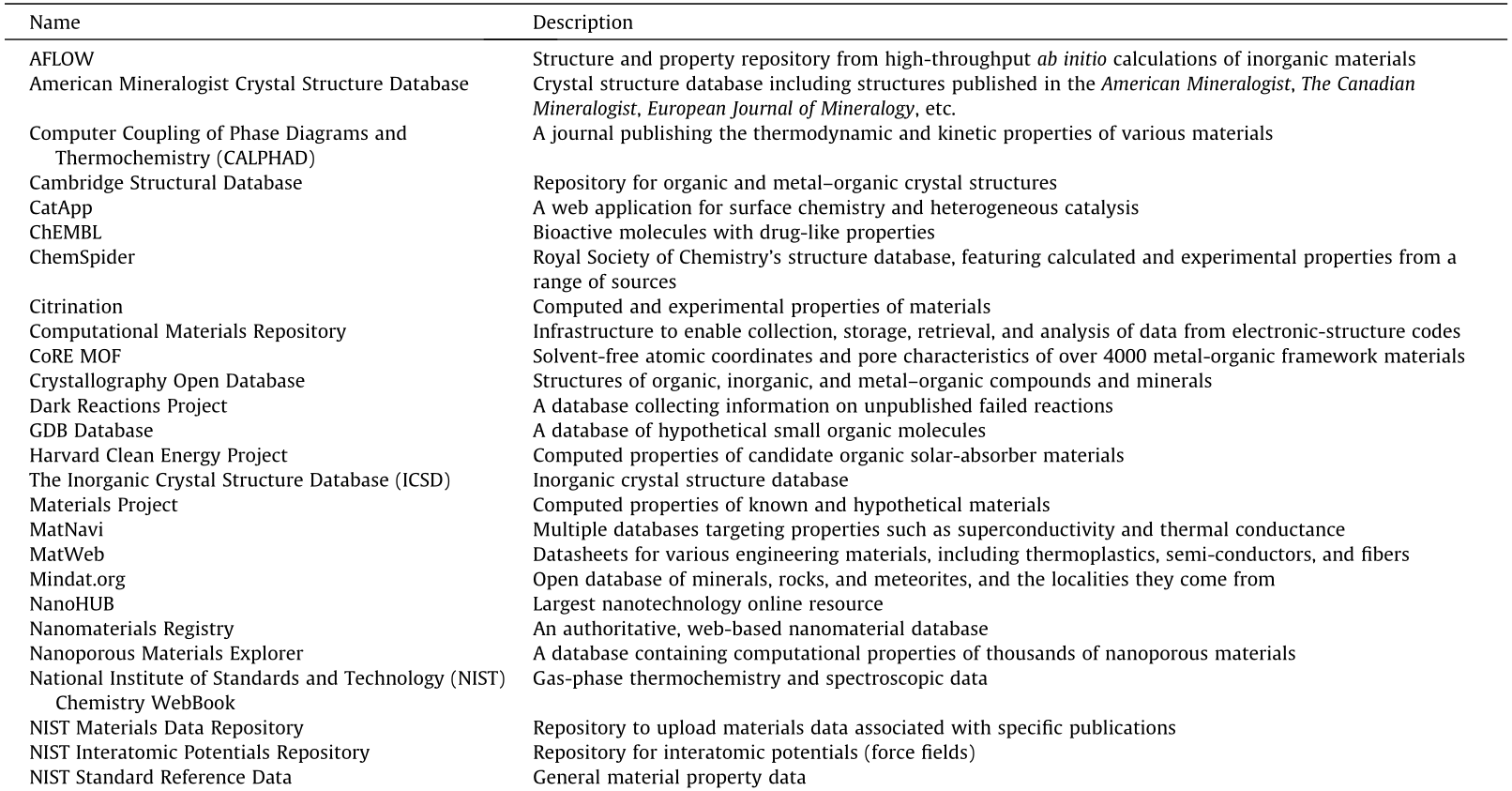
Recently, the Materials Genome Initiative (MGI) and other similar efforts around the world have been promoting the availability and accessibility of big data in materials science. There are many different kinds of materials property data (e.g., physical, chemical, mechanical, electronic, thermodynamic, and structural), which can be generated from first-principles computations (such as elastic modulus) or experimental measurements (such as thermal conductivity). Such big data has offered great opportunities for the application of data-driven techniques or ML methods to accelerate the discovery and design of new advanced materials. Table 1, updated from Ref. [18], lists many publicly available databases containing a large number of material structures and properties.
《Table 1》
Table 1 Publicly accessible structure and property databases for molecules and solid materials.

《3. ML for materials discovery and design》
3. ML for materials discovery and design
Modern theoretical and computational tools enable the efficient solving of a multitude of forward problems—that is, the prediction of the properties or behaviors of particular materials under specific conditions. Less well developed are methods and tools to handle inverse problems—that is, to design or engineer new materials with particular desirable properties. Recently, the computeraided molecular design (CAMD) method [19,20] has been proposed and significantly developed, with the aim of rationally selecting or designing molecules that possess pre-specified desirable properties. Since its emergence, the CAMD method has been used for designing solvents, pharmaceutical and consumer products, working fluids, polymers, refrigerants, and transition metal catalysts [21–31]. Similar to the CAMD problem, a typical material design task can be defined as follows: Given a {materials → property} dataset obtained from experiments and/or first-principles computations, what are the best material structure and composition that possess the most desirable properties?
For material design, the most crucial step is to build a correlation model that can accurately describe the relationship between the input material-specific features (typically structural characteristics) and the property of interest based on a given {materials → property} dataset. The construction of classical models relies heavily on physical insight and mechanisms, for example, the use of conservation laws and thermodynamics to derive mathematical formulas with parameters regressed (usually linear or slightly nonlinear) from existing reference data. ML takes a different route: Instead of relying on principles or physical insights, it trains a model with a flexible and usually highly nonlinear form solely from existing available data. In materials science, complex relationships usually exist between a material’s structure and the property of interest; these relationships are difficult to handle using traditional correlation methods. For this reason, ML methods have emerged as an important tool for predicting the properties of materials and for materials screening and optimal design.
Fig. 4 shows a general workflow for materials discovery and design based on ML. Three major steps are involved—namely, descriptor generation and dimension reduction, model construction and validation, and material prediction and experimental verification. The first step is to represent materials in the dataset numerically by a set of descriptors or features. This step requires specific domain knowledge about the materials’ class and applications. The second step is to establish a mapping model between the descriptors and the target properties based on known data for a set of reference materials. Various ML methods ranging from simple linear and nonlinear regressions to highly sophisticated kernel ridge regression and neural networks can be used to establish this mapping. In the last step, inverse design is performed to find new materials with desired properties based on the established ML models. The most promising candidates can then be synthesized and their real properties or performances can be verified experimentally.
《Fig. 4》

Fig. 4. Generic workflow for materials discovery and design based on ML.
《3.1. Descriptor generation and dimension reduction》
3.1. Descriptor generation and dimension reduction
In general, each material property depends on a set of specific factors such as crystal structure and bond strength. For this reason, the identification of key features or descriptors that are strongly correlated with the material property of interest is always a crucial step before applying the ML process. A good material descriptor should at least meet the following three criteria: It should be ① a unique characterization of the material, ② sensitive to the target property, and ③ easy to obtain. Depending on the problem or property being studied, the descriptor can be defined at different levels of complexity [32]. Taking molecular design as an example, if the boiling point or volatility of nonpolar organic compounds is being studied, the descriptor may be defined at a gross level, such as the total molecular weight. If the goal is to predict the dielectric constant, the descriptor may have to include atomic-level or at least group-level information. If catalytic activities are being investigated, the descriptor must incorporate details of the electroniclevel information.
Curtarolo et al. [33] summarized several important material descriptors that have been previously developed. The simplest descriptors are one-dimensional (1D) parameters, such as the molecular volume, weight and surface area, number of electrons, and polarities. These descriptors carry little or no information about the actual structures of the materials or molecules. As indicated before, when predicting certain properties, descriptors that represent the two-dimensional (2D) or even three-dimensional (3D) structures are preferable. Topological descriptors consider the 2D graphic structure of the molecule or material and thus reflect features such as symmetry, branching, and atom connectivity [34,35]. The most commonly used topological descriptors are the adjacency matrix [36] and the connectivity index [37]. The limitation of these descriptors is that they do not contain any stereochemistry information. An important 3D materials descriptor is the Radial Distribution Function (RDF). The RDF, which is usually denoted by g(r), defines the probability of finding a particle or atom at a distance r from another tagged particle or atom [38]. This type of descriptor can be obtained from both experimental measurements such as X-ray measurements and ab initio calculations.
A substantial number of databases (see Table 1) contain a large amount of material structure and property data. However, it should be noted that the available materials data are often highly correlated to each other. Therefore, it is necessary in many cases to pre-process the high-dimensional datasets with dimension reduction tools prior to the construction of ML models. Several algorithms [39] such as principal component analysis (PCA), multidimensional scaling (MDS), and linear discriminant analysis (LDA) are available to reduce the dimension of the feature space and help identify the most relevant descriptors (or key features) for ML. For example, PCA converts a set of correlated variables into a reduced set of uncorrelated new variables or principal components (PCs) using orthogonal transformation [40]. Each PC is chosen so that it lies along the direction of largest variance while being uncorrelated to other PCs. The PCs constitute a reduced dimensional space that can represent the original data with a limited loss of information. Zhou et al. [41] employed PCA to reduce a 12-dimensional solvent descriptor space to a four-dimensional space. The four new descriptors were then successfully used to correlate and predict solvent effects on reaction rates.
《3.2. Model construction and validation》
3.2. Model construction and validation
ML algorithms can be broadly classified into two categories: supervised and unsupervised learning algorithms. Supervised learning may be further classified into regression and classification. In materials design, supervised learning attempts to identify a function that is able to predict the properties for new materials based on a set of known materials and their properties. If the target property is a continuous quantity (e.g., glass transition temperature), the process is known as regression. Typical regression algorithms are Kriging or Gaussian process regression [42], artificial neural networks (ANNs) [43], and support vector machines (SVMs) [44]. If the outputs are discrete targets (e.g., whether toxic or not, which type of crystal), the process of searching for the prediction function is then known as classification. The decision tree [45] and random forest [46] algorithms are the two most commonly used classification algorithms.
While supervised learning aims to find a function mapping a set of input data to a corresponding output property, unsupervised learning attempts to identify the relationship among the input data themselves. Clustering, as a typical unsupervised learning method, is the process of partitioning a dataset into different categories or regions, such that the data points in the same group or cluster are more similar to each other than to those in other clusters. Clustering is very useful for extracting physical insights from data and for finding new promising materials based on comparative studies [47]. The most popular clustering algorithms are k-means [48], hierarchical clustering [49], and hidden Markov modeling [50].
A list of important ML methods is summarized in Table 2, and a detailed introduction to each method can be found in Ref. [51]. Since each method or algorithm has its own suitability and application scope, the selection of an appropriate ML algorithm is crucial for its successful implementation. Several algorithms, such as least-squares regression, kernel ridge regression, neural networks, and decision trees, are able to create property prediction models. However, while some algorithms (mainly regression-based ones) provide the actual predictive functions, others (e.g., decision trees) do not. Moreover, the amount of available data can also dictate the selection of learning algorithms. For example, tens to thousands of data points may be properly handled with regression methods such as Kriging and kernel ridge regression. However, when the dataset is much larger, more sophisticated learning methods such as deep neural networks should be applied [32].
《Table 2》
Table 2 A list of important ML methods.

In recent years, many open-source software programs or tools such as scikit-learn, TensorFlow, and Chainer have been developed, making it possible for non-specialists to implement ML methods within their own research. Scikit-learn is a Python package that integrates a wide range of state-of-the-art ML algorithms, both supervised and unsupervised. TensorFlow is a software library for high-performance numerical computation. Originally developed by researchers and engineers from Google’s AI department, TensorFlow now provides strong support for ML and deep learning. Chainer is a powerful tool for constructing neural networks, which aims to bridge the gap between algorithms and implementations. The commercial software MATLAB also incorporates many ML algorithms in toolboxes such as Statistics.
A data-driven model can, in principle, memorize every data point in the training set and thus result in extremely high accuracy regarding these data. For this reason, ML models must be evaluated on data that have not been used for training. The simplest way is to perform cross validation, where the model is built on only part of the data and the remaining data is used for evaluation or validation. There are several cross-validation strategies, among which the k-fold cross-validation method [52] is very popular. In this strategy, a dataset is randomly partitioned into k subgroups with the same size; the (k – 1) subsamples are used for training and the remaining one subsample is used for validation. This crossvalidation process is repeated k times, with each of the k subsamples used exactly once as validation data. Kohavi [53] demonstrated that for real-world datasets, the best method for model validation is ten-fold cross-validation, even though computational power permits the use of more folds. Another widely used method for validating ML models is the bootstrap method [54]. Here, a “bootstrap training set” with the same size as the original dataset is constructed by extracting samples from the original dataset one at a time and returning them back to the dataset after they have been chosen. As a result, some data points may appear more than once in a bootstrap training set, while others may not appear at all. The data points that have not been used in the training set are then used for model validation. The above procedure can be repeated several times, and the averaged prediction error is used as an indicator of the model performance. An advantage of the bootstrap method is that the result can be presented with confidence intervals or uncertainties—a feature not readily available with other validation methods.
《3.3. Material prediction and experimental verification》
3.3. Material prediction and experimental verification
As indicated in Fig. 4, after the ML model is established, inverse design can be performed to find materials with desired properties based on the model. This can be done by using either large-scale screening or mathematical optimization.
The basic idea of the large-scale screening method is to first generate all possible material candidates in the design space, and then test them one by one using the learned model [15]. Typically, the generation of materials must consider several constraints on the representation of the material, which is normally in the form of a structure and/or composition-based function. For this reason, a systematic procedure is required to identify all the materials (or as many as possible) in the design space. Once the candidates are generated, the evaluation of their properties is simple and straightforward with the trained model.
Alternatively, reverse materials design can be formulated as a mathematical optimization problem, where the target property is optimized subjected to the structural and composition constraints [55,56]. The optimization-based method attempts to identify promising materials without testing all the candidates in the design space. This feature makes the method much less limited by the combinatorial complexity. Either deterministic [57] or stochastic algorithms [58] can be employed to solve the formulated optimization problem for which the optimal materials are identified.
After the best materials are identified, they can be synthesized and their actual properties can be experimentally verified. If the experimental results agree well with the predicted ones, the materials are confirmed to have the highest performance. If not, the designed materials and corresponding experimental results are added to the training set and the ML model is retrained.
《4. Application examples》
4. Application examples
ML has accelerated the development of several different kinds of materials. In this review article, we have chosen to focus on three classes of materials: polymer and porous materials, catalytic materials, and energetic materials. Recent applications of ML methods for the discovery and optimal design of these materials are highlighted in the following sections.
《4.1. Polymer and porous materials》
4.1. Polymer and porous materials
Polymer materials have many desirable properties such as a high strength-to-weight ratio, resistance to corrosion, being easy to shape, and having a low manufacturing cost. Due to these advantages, polymer materials are finding increasing applications in many engineering areas, from traditional packaging and consumer products to electrochemical and biomedical engineering. Based on the large quantity of existing polymeric structural and property data, the data-driven or ML method can play an important role in polymer discovery and design.
Breneman et al. [59] developed a materials genomics approach for the optimal design of spherical nanoparticle-filled polymers based on the prediction of their thermomechanical properties. Experimental studies were used to validate the design results. Venkatraman and Alsberg [60] proposed an ML model to rapidly discover new polymer materials with multiple desirable properties including a high refractive index. The obtained results were successfully verified by means of DFT calculations. To facilitate the development of new polymer materials, Wu et al. [61] established statistical models to predict the dielectric constant, band gap, dielectric loss tangent, and glass transition temperature for organic polymers. A new set of features called infinite chain descriptors was developed to characterize organic polymers, and was used as inputs for ML to predict the aforementioned properties. It was found that all the obtained ML models showed good performance in polymer property predictions. Sukumar et al. [62] demonstrated how to build ML models for the optimal design of polymers with specific electronic properties. Model validation confirmed that the established models were able to make reliable predictions on polymers outside of the training set.
Dielectric materials are traditionally made from inorganic materials such as porcelain, mica, and quartz. However, when used as dielectric materials, polymers provide the advantages of excellent chemical resistance, flexibility, cheapness, and tunability for specific applications. Sharma et al. [11] proposed a hierarchical ML-based method to accelerate the identification of polymer dielectrics that outperform standard materials. The measured dielectric properties for some of the designed polymers strongly support the efficacy of the proposed approach for optimal polymer dielectrics design. Mannodi-Kanakkithodi et al. [56] performed polymer dielectrics design by building statistical learning models based on data generated from first-principles computations. The polymers were fingerprinted as simple numerical representations, which were then mapped to the properties of interest using an ML algorithm. Moreover, a genetic algorithm was used to optimize polymer constituent blocks in an evolutionary manner, thus directly leading to the design of polymers with the target properties. Through the development of a polymer genome, MannodiKanakkithodi et al. [63] also presented an essential roadmap for the design of polymer dielectrics, along with future directions for expansions to other polymer classes and properties.
Metal–organic frameworks (MOFs), as an important porous material, possess great potential for many applications, and particularly for gas storage and separation. Furthermore, the structural building blocks of MOFs can be combined to synthesize a nearly infinite number of materials. This makes computational methods very useful for the large-scale screening and optimal design of MOF materials.
Fernandez et al. [64] reported the first QSPR analysis of MOFs for methane (CH4) storage. These scholars investigated the effect of geometrical features—namely, pore size, surface area, and void fraction—as well as the framework density on the simulated CH4 storage capacities of about 1.3 ×105 hypothetical MOFs at 1, 35, and 100 bar (1 bar = 100 kPa). Based on these data, several ML models including multi-linear regression models, decision trees, and nonlinear SVMs were developed to predict the CH4 storage capacities of MOFs. In each case, 1 ×104 MOFs were used to train the model, and the accuracy of the model was validated on a test set of about 1.2×105 MOFs. It was found that for CH4 storage at 35 bar, desirable MOFs should have densities greater than 0.43 g·cm-3 and void fractions greater than 0.52; for CH4 storage at 100 bar, MOFs should have densities greater than 0.33 g·cm-3 and void fractions greater than 0.62. Based on the response surface analyses of the SVM model, the researchers identified new materials that might lead to extremely high CH4 storage capacities. In order to accurately predict carbon dioxide (CO2) uptakes in MOFs, Fernandez et al. [65] introduced the atomic property-weighted RDF (AP-RDF) descriptor, which captures the chemical features of a periodic material in addition to its geometric features. Nonlinear SVM models based on the AP-RDF descriptors yielded good predictions on CO2 equilibrium loadings at both 0.15 and 1 bar. This result suggests that MOFs with more compact frameworks and interatomic distances in the range from 6 to 9 Å exhibit a higher affinity for CO2 at both pressures. Ohno and Mukae [66] applied Gaussian process regression to correlate and predict the equilibrium CH4 loading of MOFs. Based on the established model, optimal MOFs that could outperform all the materials in the model training set were successfully identified.
Aghaji et al. [10] employed decision tree and SVM methods to predict the CO2 uptake capacity and CO2/CH4 separation selectivity of MOFs by using geometrical descriptors of the materials as the ML input variables. It was found that pore size, void fraction, and surface area were the most important factors for designing optimal MOFs for separating CO2 from CH4. Simon et al. [67] used the random forest method to discover new porous materials with great potential for xenon and krypton separation. Two high-performing materials were identified: an aluminophosphate zeolite analogue and a calcium-based coordination network. Both materials have been synthesized, but they have not yet been tested for xenon and krypton separation. Fernandez and Barnard [68] developed ML models for predicting the CO2 and nitrogen (N2) uptake capacities of MOFs. Many different ML techniques, including the decision tree, k-nearest neighbor, SVM, ANN, and random forest methods, were investigated. It was found that the random forest method yielded the most accurate predictions on both CO2 and N2 uptake capacities. Based on the established models, the most promising MOFs for efficient CO2/N2 separations were identified. Qiao et al. [69] applied the decision tree method to study the relationship between the geometrical descriptors of MOFs and the MOF-membrane performance for separating a ternary gas mixture (CO2/N2/CH4) at 298 K and 10 bar. The seven best MOF membranes were finally identified.
《4.2. Catalytic materials》
4.2. Catalytic materials
Catalysts are used in many industrial processes. Traditionally, the optimal design of catalysts has been empirical or has mostly depended on experimentation. Quantum chemical calculations provide the possibility for first-principles catalyst design. However, the large computational cost limits their application to relatively simple reactions and to a small number of catalyst candidates. With the rapidly increasing amount of available experimental and computational data, as well as the development of catalysis informatics, catalyst structure and activity relationships can now be well described using ML models, which are very useful for catalyst development.
One of the first attempts to use ML methods for catalyst design was carried out by Huang et al. [70], who developed an ANN model to describe the relation between catalyst components and catalytic performance. A hybrid genetic algorithm was proposed and used to find the optimal multicomponent catalysts based on a trained ANN model. The catalyst design strategy was successfully applied to the CH4 oxidative coupling reaction. A few high-performance catalysts were found, and the C2 hydrocarbon yield of the best catalyst reached 27.78%, which was higher than the yields of previously reported catalysts. Baumes et al. [71] employed ANN models to predict catalyst performance for the water gas shift reaction. It has been shown that compared with traditional computational and experimental trial-and-error approaches, ML methods possess great potential for accelerating the discovery of high-performance heterogeneous catalysts. Baumes et al. [72] introduced linear SVM models to optimize olefin epoxidation catalysts. Later, a nonlinear SVM model was trained for a second catalytic reaction—that is, light paraffin isomerization. Based on these two application examples, the researchers discussed the advantages of SVM for catalyst research in comparison with other ML techniques such as neural networks and decision trees.
Thornton et al. [73] developed an ML model for the computational screening of over 3 ×105 zeolite catalysts for CO2 reduction. It was found that an optimal cavity size of around 6 Å is required to maximize the change in entropy–enthalpy upon adsorption with a maximum void space greater than 30% to promote product formation. Corma et al. [74] described how spectral characterization descriptors can be used in combination with conventional structural and composition descriptors for the construction of catalyst performance prediction models. PCA was first employed to extract the desired spectral descriptors from the X-ray diffraction (XRD) characterization of the catalyst. Performance prediction models were then obtained by using ANN and decision tree modeling techniques. Through the application to an epoxidation reaction based on mesoporous titanium (Ti)-silicate catalysts, it was demonstrated that the use of spectral descriptors can remarkably increase the prediction accuracy of the ML model and thus improve the reliability of the catalyst design results. Mixed metal oxides are robust materials that are often used as industrial catalysts. However, predicting their catalytic performance a priori is difficult. Using the oxidative dehydrogenation of butane to 1,3-butadiene as a model reaction, Madaan et al. [12] experimentally synthesized and tested 15 mixed bimetallic oxides supported on alumina. Based on the experimental results, a descriptor model was built and used to predict the performance of a set of 1711 mixed-metal oxide catalysts. Six new promising bimetallic oxide catalysts were identified and experimentally verified.
Bimetallic and multi-metallic catalysts exhibit high activities for a wide range of thermal and electrochemical reactions. However, modeling the many diverse active sites is a significant challenge. Li et al. [75] developed ML models for the rapid screening of transition-metal catalysts using easily accessible catalyst descriptors as the model inputs. The descriptors include the local electronegativity and effective coordination number of an adsorption site, as well as intrinsic properties of active metal atoms, such as the ionic potential and electron affinity. The trained models were used to screen multi-metallic alloys for electrochemical CO2 reduction. Several promising catalyst candidates were identified. Li et al. [76] presented an ANN-based framework for the rapid screening of bimetallic catalysts using methanol electro-oxidation as the model reaction. A catalyst database containing the adsorption energies of *CO and *OH on {111}-terminated model alloy surfaces and fingerprint features of active sites from DFT calculations was established and used to optimize the structural and weight parameters of the ANN. The fingerprint descriptors include the sp-band and d-band characteristics of an adsorption site together with tabulated properties of host-metal atoms. It was demonstrated that an ANN model trained with the existing dataset of about 1000 idealized alloy surfaces could capture the complex adsorbate/metal interactions, and showed high predictive power in exploring the large chemical space of bimetallic catalysts. Ulissi et al. [77] proposed another framework for designing bimetallic catalysts. Active sites for every stable low index facet of a bimetallic crystal were enumerated and cataloged, yielding hundreds of possible active sites. The activities of these sites were predicted in parallel using an ANN-based surrogate model. Sites with high activities were found, which provided targets for subsequent DFT calculations. The design framework was applied to the electrochemical reduction of CO2 on nickel gallium bimetallics.
Nanomaterial-based catalysts are usually heterogeneous catalysts broken up into metal nanoparticles. Metal nanoparticles have larger surface areas than their bulk counterparts, so their use results in increased catalytic activity [78]. Fernandez et al. [79] developed decision tree and ANN models to predict the catalytic activities of platinum nanoparticles from their structural descriptors such as particle diameter, surface area, and sphericity based on a dataset derived from DFT calculations. It was demonstrated that ML techniques can be used to rapidly estimate the catalytic properties of nanomaterials at a resolution that is inaccessible to both experimental and ab initio methods. Principles or rules for guiding the rational design of nanocatalysts in the near future were identified. As is widely known, catalytic activities are normally dominated by a few specific surface sites. Therefore, designing active sites is the key to the realization of high-performance heterogeneous catalysts. Alloy nanoparticles have a distribution of active sites that may differ from those on single-crystal surfaces. This makes the optimal design of alloy nanoparticles very challenging. Jinnouchi and Asahi [8] proposed an ML scheme using a local similarity kernel, which makes it possible to understand and approximate the catalytic activities of alloy nanoparticles based on local atomic configurations. This method has been successfully applied to the direct NO decomposition reaction on Rh-Au alloy nanoparticles.
Data-driven modeling is not only important for heterogeneous catalyst design, but also for homogenous catalysis. Maldonado and Rothenberg [80] summarized why, when, and how predictive modeling should be used for homogenous catalyst design. Transition metal complexes, which are a type of important homogeneous catalyst, have very complex electronic structures, and direct DFT simulation of these materials is very computationally expensive. Janet and Kulik [81] used ANN methods to predict the electronic properties for transition metal complexes including spin-state ordering and specific bond lengths. It was shown that the ANN outperformed other ML methods, including SVM and kernel ridge regression. The developed ANN model provides a good basis for the large-scale screening of transition metal complex catalysts.
《4.3. Energetic materials》
4.3. Energetic materials
ML plays an important role in accelerating the discovery of high-performance energetic materials, including battery and superconductor materials, electroceramic and thermoelectric materials, and photovoltaic and perovskite materials.
Fujimura et al. [82] used ML methods to predict the conductivity of different compositions of lithium (Li)-conducting oxides as Li-ion materials at 373 K based on experimental and computational data. Rational design of superior Li-ion conductors was performed by optimizing the materials’ compositions based on the established ML models. Crystal structures have a great impact on the physical and chemical properties of Li-ion silicate cathodes and thus greatly influence their battery applications. Three major crystal types (i.e., monoclinic, orthorhombic, and triclinic) of silicate-based cathodes were predicted by Shandiz and Gauvin [83] using different classification algorithms. It was demonstrated that the random forest method yielded the highest prediction accuracy in comparison with other classification methods. Sendek et al. [84] presented a large-scale computational screening approach for identifying promising candidate materials for solid-state electrolytes for Li-ion batteries. The authors first screened 12831 Li-containing crystalline solids with high structural and chemical stability, low electronic conductivity, and low cost. They then developed a data-driven ionic conductivity classification model using logistic regression to further select candidate structures that exhibit fast Li conduction. The number of candidate materials was reduced from 12831 to 21, a few of which have been examined experimentally. Stanev et al. [14] used several ML schemes to develop different models to predict the critical temperatures of more than 1.2 ×104 superconductors. To improve the accuracy and interpretability of these models, new descriptors were incorporated using materials data from the AFLOW Online Repositories. Finally, the regression and classification models were combined into a single pipeline, which was employed to search the entire Inorganic Crystal Structure Database (ICSD) to find potential new superconductors with desirable critical temperatures. More than 30 non-cuprate and non-iron-based oxides were successfully identified.
Scott et al. [85] used ANN methods to design electroceramic materials based on a recently established database containing composition and property information for a wide range of ceramic compounds. A stochastic optimization algorithm was employed to search for optimal materials considering the properties of high relative permittivity and low overall charge. It was found that in some cases, the identified materials were similar to those contained in the database; in other cases, completely new materials were found. Based on available knowledge on 2.5 ×104 known materials, Gaultois et al. [86] developed an open-source ML-based engine for the evaluation of the performance of thermoelectric materials. It was demonstrated that this engine can identify promising thermoelectric materials that are different from known ones.
The growth in energy demands coupled with the need for clean energy is likely to make solar cells an important energy supplier. Photovoltaic and perovskite materials are two of the main materials for the storage and utilization of solar energy. Nagasawa et al. [87] screened conjugated molecules for organic photovoltaic applications by using ANN and random forest modeling. Parameters including the molecular weight, electronic property, and power conversion efficiency were collected from the literature and subjected to ML. It was demonstrated that the random forest model yielded higher prediction accuracy than the ANN-based model. Olivares-Amaya et al. [15] used ML techniques to develop models for predicting important current-voltage and efficiency properties of potential organic photovoltaic molecules. The obtained models were used to quickly screen promising photovoltaic materials from 2.6 million candidate compounds. The results revealed that the benzothiadiazole and thienopyrrole homologues are currently the most promising set of molecules for photovoltaic applications. Yosipof et al. [88] proposed a data mining and ML workflow, and applied it to the analysis of two recently developed solar cell libraries based on Ti and copper oxides. The results demonstrated that the ML model built from the k-nearest neighbor algorithm can yield good predictions for multiple solar cell properties. This model is therefore suitable for designing better photovoltaic solar cells based on new promising metal oxides.
A perovskite solar cell is another type of solar cell that includes a perovskite-structured compound—most commonly a hybrid organic–inorganic lead or tin halide-based material—as the lightharvesting active layer [89]. Accurate prediction of the bandgaps of double perovskites is significant for their solar cell applications. While quantum mechanical computations for quantifying bandgaps are very computationally expensive, data-driven ML approaches are promising alternatives. Pilania et al. [7] developed a robust ML framework for the efficient and accurate prediction of the electronic bandgaps of double perovskites. The established learning models were validated and used to design promising perovskite materials for solar cell applications. Curie temperature (Tc), the second-order phase-transition temperature, is another important physical property for perovskite materials. Zhai et al. [55] employed the SVM, relevance vector machine, and random forest methods to establish prediction models for Tc. According to the k-fold cross-validation, the SVM model shows better prediction performance than the other two models. Potential perovskite materials with high Tc were found based on the SVM model using a genetic-algorithm-guided search strategy.
《5. Conclusion》
5. Conclusion
Data-driven science, the fourth paradigm of science, has given rise to the MGI and to materials informatics. The progress of the MGI and materials informatics has totally changed the philosophy of materials research and development. Instead of relying on experimental trial-and-error or high-throughput ab initio calculations, data-driven or ML methods are now playing significant roles in predicting the properties of various materials and guiding experimentalists to discover and develop new high-performance materials. This review article provided a brief introduction on different classes of ML algorithms as well as related software and tools. The basic steps for applying ML methods for materials discovery and design were summarized. Recent applications on the largescale screening and rational design of polymer and porous materials, catalytic materials, and energetic materials were highlighted. Despite a substantial number of successful applications, this exciting topic is still largely in its nascent stage and it is believed that ML will play an increasingly important role in accelerating the development of various kinds of functional materials in the foreseeable future.
《Acknowledgement》
Acknowledgement
The authors acknowledge the financial support from Max Planck Society, Germany.
《Compliance with ethics guidelines》
Compliance with ethics guidelines
Teng Zhou, Zhen Song, and Kai Sundmacher declare that they have no conflict of interest or financial conflicts to disclose.











 京公网安备 11010502051620号
京公网安备 11010502051620号




