

《1. Introduction》
1. Introduction
In December 2019, a series of pneumonia cases of unknown cause emerged, followed by a rapidly spread due to strong human-to-human transmission [1]. Based on the clinical presentation, the pneumonia was determined to be a viral infection; the virus was initially named the 2019 novel coronavirus (2019- nCoV) and then, formally, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [2]. The World Health Organization (WHO) declared COVID-19 to be a public health emergency of international concern on 30 January 2020 [3]. The mortality rate was found to be 3.9% according to the data at that time [4]. A number of studies and reports have identified a median incubation period of 4 d and have determined that the top four symptoms include fever, cough, shortness of breath, and chest tightness/pain [5–7]. The most unfortunate fact is that there have been no effective therapies for preventing and treating COVID-19 to date. Although remdesivir and hydroxychloroquine have been found to be effective in inhibiting SARS-CoV-2, the data obtained thus far are primarily from in vitro studies [8]. Interferon, lopinavir/ritonavir, arbidol, ribavirin, and the therapeutic application of plasma antibodies have also been recommended as alternatives for the treatment of patients with COVID-19; however, the efficacy and safety of these drugs remain to be verified in patients, and their applications are yet to be validated by scientifically sound randomized clinical trials (RCTs) [9,10].
Triazavirin (TZV), a new antiviral drug, has been on the market in Russia since 2015. It is a synthetic compound analogue to the purine nucleoside bases. The principle mode of action of TZV is inhibiting the synthesis of viral RNA and preventing the replication of genomic fragments [11]. Because of its multiple-target mechanism of action, TZV has a wide spectrum of antiviral activity against RNA-containing viruses, including influenza A virus (H5N1, etc.), influenza B virus, tick-borne encephalitis, and Forest-Spring encephalitis, both in vitro and in animal models in vivo [11–13]. According to the package insert, the recommended dosage of TZV is 250 mg three times daily, for a consecutive 5–7 d. In a phase II RCT on TZV, patients took 250 mg TZV orally three times a day for 5 d; the results showed that TZV can significantly shorten the duration of the major clinical symptoms of influenza and decrease the incidence of influenza-related complications and complications induced by the concurrent use of symptomatic drugs. No obvious adverse events (AEs) were reported [14,15]. Nevertheless, the efficacy and safety of TZV for COVID-19 remain uncertain until the anticipated positive results are obtained from RCTs. We therefore set up the present ongoing multicenter and blind RCT with the aim of testing the efficacy and safety of TZV for COVID-19.
《2. Methods》
2. Methods
《2.1. Study design》
2.1. Study design
This clinical study is an ongoing multicenter and blind RCT to demonstrate the efficacy and safety of TZV versus a placebo in the treatment of COVID-19. Patients with positive nucleic acid tests for SARS-CoV-2 are randomly assigned into two groups in equal proportions (1:1). The trial group receives TZV plus standard treatment according to the Diagnosis and treatment protocol for novel coronavirus pneumonia (trial version 7) and the Diagnosis and treatment protocol for novel coronavirus pneumonia (trial version 5) drafted by the National Health Commission of the People’s Republic of China and the National Administration of Traditional Chinese Medicine [9,16], and the placebo group receives the TZV placebo plus standard treatment. Patients undergo consecutive 7- day treatment and are then followed up on Days 3, 7, 14, and 21 after the end of treatment. Specific items that will be recorded for each intervention and follow-up period are outlined in Table 1. This trial is registered on the Chinese Clinical Trial Registry↑ with the identifier number ChiCTR20000300001.
↑ http://www.chictr.org.cn/.
《Table 1 》
Table 1 Standard protocol items and procedures.
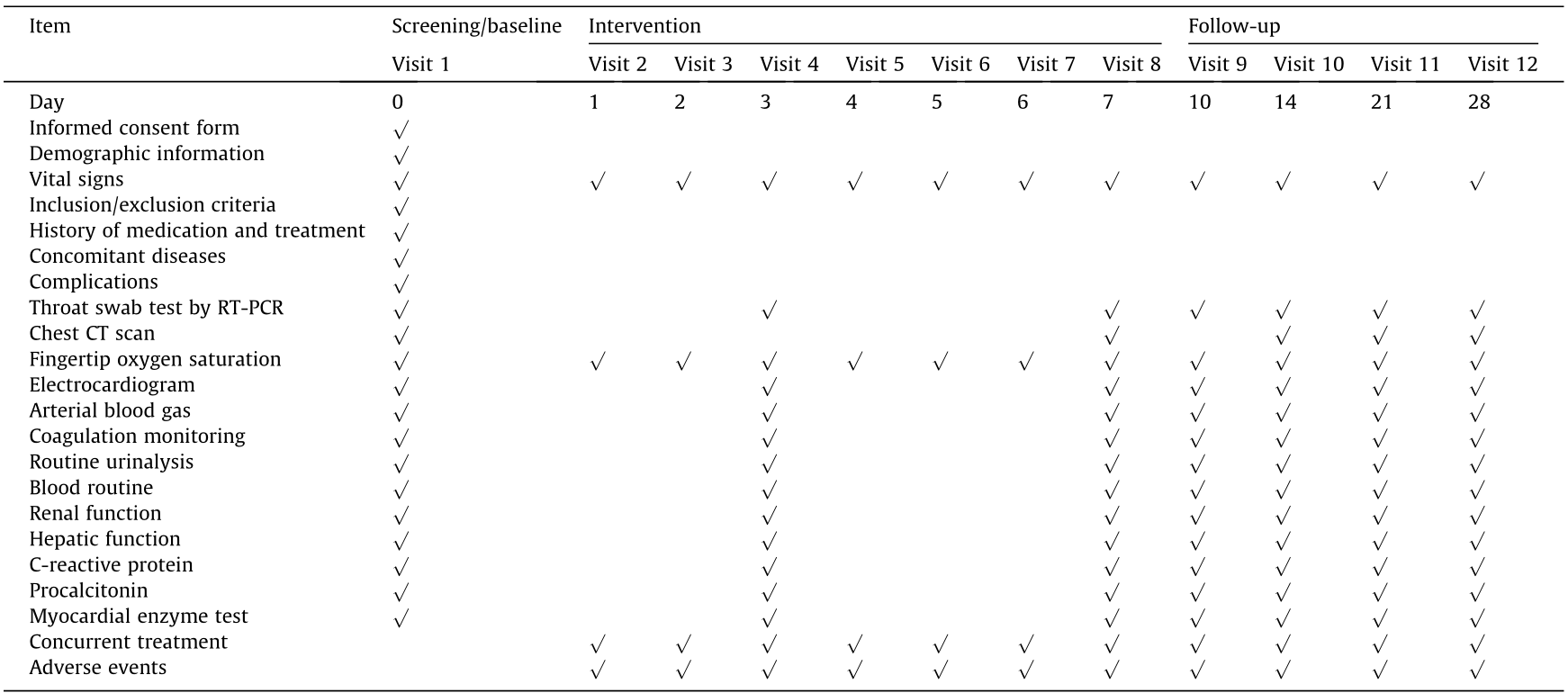
RT-PCR: reverse transcription polymerase chain reaction; CT: computed tomography.
《2.2. Study setting》
2.2. Study setting
The ten hospitals enrolled in this study are all officially ranked as Level-3 Class-A medical centers with recognized qualifications and credentials (Table 2). The study was initiated on 14 February 2020, and the planned date for the completion of the study is 31 May 2020.
《2.3. Ethics》
2.3. Ethics
This clinical trial is being carried out according to the principles of the Declaration of Helsinki‡ and follows the laws, regulations, and administrative provisions of the National Health Commission of the People’s Republic of China↑↑. The trial was commenced after approval was obtained from the ethic committees (detailed information is listed in Table 2). Participants are informed of the risks and benefits of the study, and are allowed to voluntarily cease participation in the study at any time, for any reason. To protect the privacy of subjects, each patient is identified with a unique random number, and patients’ names and personal information are kept confidential to everyone except for the researchers.
‡ https://www.wma.net/what-we-do/medical-ethics/declaration-of-helsinki/.
↑↑ http://www.nhc.gov.cn/yzygj/s7659/202004/1d5d7ea301f04adba4c4e47d2e92eb96.shtml.
《Table 2》
Table 2 Study settings and name of each ethics committee.

《2.4. Eligibility criteria》
2.4. Eligibility criteria
Patients are being enrolled from the emergency departments, isolation wards, and intensive care unit (ICU) departments of the ten study sites. A flow chart of the study is shown in Fig. 1. In brief, patients with a definite diagnosis of COVID-19 (confirmed positive by laboratory real-time reverse transcription polymerase chain reaction (RT-PCR) are screened. Patients meeting all inclusion criteria and no exclusion criteria (to be specified below) are candidates for this study. A total of 24 participants are scheduled to be enrolled at each study site, resulting in a total of 240 participants in the study.
《Fig. 1》
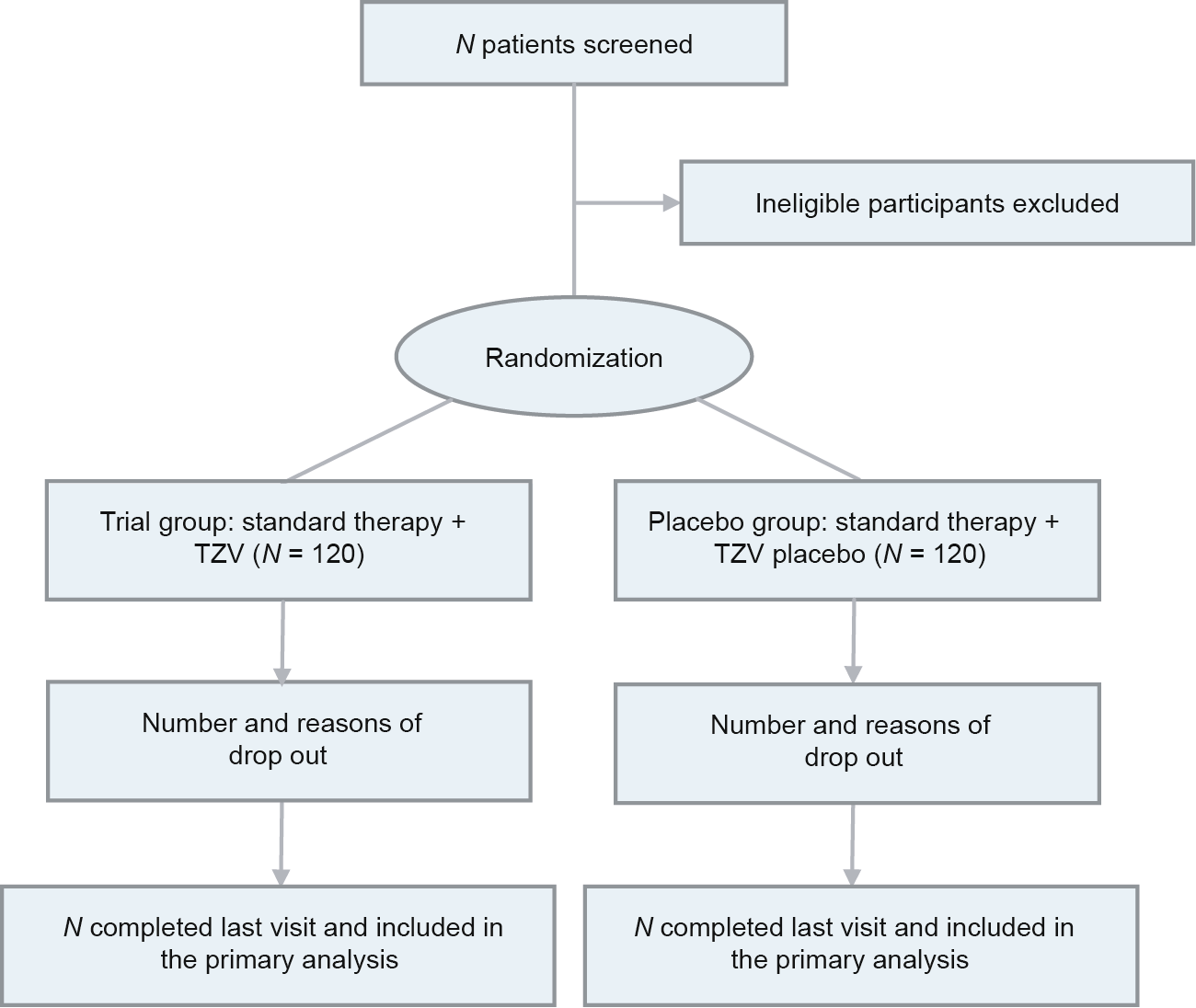
Fig. 1. Flow chart of the study.
The inclusion criteria are as follows: ① adults more than 18 years old who provide informed consent; ② laboratoryconfirmed SARS-CoV-2 infection by real-time RT-PCR; ③ chest computed tomography (CT) imaging-confirmed lung damage, including multiple small plaques and stromal changes in the lungs, which are obvious in the outer lung, or multiple ground-glass shadows and infiltration shadows in both lungs, although these changes might not be present in mild patients; ④ hospitalized patients with fever (axillary temperature ≥37.0 °C) or respiratory symptoms; ⑤ an interval between symptom onset and randomization of ≤ 12 d; ⑥ no participation in other clinical research in the past three months; and ⑦ no participation in other antiviral studies within the entire study period of 28 d.
The exclusion criteria are as follows: ① patients who are unsuitable or who cannot participate safely in the study, as judged by the principle investigators (PIs); ② patients with serious liver disease of grade C according to the Child–Pugh score; ③ patients with severe renal impairment (estimated glomerular filtration rate ≤30 mL·min-1 ·1.73 m-2 ) or continuous renal replacement therapy, hemodialysis, or peritoneal dialysis; ④ patients with severe anemia (hemoglobin (Hgb) < 60 g·L–1); ⑤ women with a positive pregnancy test, ongoing pregnancy, or who are breastfeeding; ⑥ patients with a history of allergy to TZV or its metabolic components; ⑦ patients who have not provided informed consent; ⑧ patients who have a possibility of being transferred to another hospital within 72 h of randomization; and ⑨ patients participating in other clinical trials for COVID-19 within 30 d prior to screening.
Patients may withdraw, discontinue, or drop out from the trial at any time. Data recorded up to the point of withdrawal will be included in the trial analysis. The withdrawal/drop-out criteria are as follows: ① voluntary withdrawal; ② poor compliance with the intervention, such that patients receive treatment for fewer than 3 d; ③ inability to take the drug because of deterioration of conditions, occurrence of serious adverse events (SAEs), complications, fatal physiological changes, or even death, and a high possibility of withdrawal from the trial as judged by the investigators; ④ patients for whom clinical data collection and statistical analyses after inclusion are difficult to conduct; and ⑤ patients who are taking medications that have a definite interaction with the current study drug.
The trial discontinuation criteria are as follows: ① major deviation from or violation of the study protocol, making it difficult to evaluate the efficacy and safety of the drug; ② serious safety problems found during the study; ③ no clinical effects observed during the study, so as to avoid delayed treatment of patients and unnecessary economic burden; ④ the sponsor proposes the termination of the study to protect the rights and security of the subjects; or ⑤ termination of the study by the decision of the Data and Safety Monitoring Board (DSMB) handling the interim analysis.
《2.5. Intervention and study visits》
2.5. Intervention and study visits
2.5.1. Study procedures
The treatment period is 7 d, the follow-up period is 21 d, and the entire study period is 28 d. The planned specific visits and measurements are summarized in Table 1.
2.5.2. Interventions
The trial group receives standard therapy and TZV (250 mg at a time, three times a day for 7 d in mild or ordinary cases, and four times a day for 7 d in severe or critically severe cases). The placebo group receives the same standard therapy plus a TZV placebo (250 mg at a time, three times a day for 7 d in mild or ordinary cases, and four times a day for 7 d in severe or critically severe cases). The first dose of the study drug is administered immediately after randomization. The TZV is manufactured by Medsintez (Russia). The TZV and placebo have been repackaged and labeled in the same way by the Tianqing Stem Cell Co., Ltd. (China), following the applicable Good Manufacturing Practices. The TZV placebo capsule contains pharmaceutical excipients only, and has the same appearance and smell as the TZV.
A patient’s condition is considered mild if the clinical symptoms are mild and no pneumonia manifestation can be found in chest CT imaging. A patient’s condition is considered ordinary if symptoms such as fever and respiratory tract symptoms are present, and if signs of pneumonia can be seen on imaging. A severe condition is defined as a respiratory rate ≥30 breaths per minute, pulse oxygen saturation (SpO2) ≤93% in room air at a rest state, arterial partial pressure of oxygen (PaO2)/fraction of inspiration oxygen (FiO2) ≤300 mmHg (1 mmHg = 133.3 Pa), or > 50% lesions progression within 24–48 h in pulmonary imaging. A critically severe condition is defined as respiratory failure resulting in mechanical ventilation, the occurrence of shock, or the occurrence of other organ failure requiring monitoring and treatment in the ICU.
2.5.3. Screening visits
Screening begins 24 h before recruitment, and baseline data are obtained during the screening period. Examinations and laboratory tests are all performed at the local study sites, including routine monitoring of body vital signs, fingertip oxygen saturation, electrocardiogram, arterial blood gas, routine urinalysis, routine blood test, C-reactive protein, hepatic and renal functions, coagulation monitoring, procalcitonin, myocardial enzyme test, chest CT imaging, and throat swab test by laboratory RT-PCR.
2.5.4. Intervention visits
The intervention period is 7 d. Clinical symptoms, routine monitoring of vital signs, fingertip oxygen saturation, concurrent medications, and AEs are recorded on a daily basis. Electrocardiograms and other laboratory tests are performed at Days 3 and 7.
2.5.5. Follow-up visits
A 21-day follow-up of the patients is conducted. If the patient is in the hospital, clinical symptoms, routine monitoring of vital signs, fingertip oxygen saturation, concurrent medication, and AEs are recorded every day. Electrocardiograms and other laboratory tests are performed at Days 10, 14, 21, and 28. For patients who are discharged from the hospital during the follow-up period, concurrent medications and AEs are recorded by telephone followup.
《2.6. Randomization》
2.6. Randomization
Block randomization with a block size of four was conducted and stratified by having the research at every study site once the eligibility requirements have been met. A placebo-control blind method was used in this study. Participants fulfilling the inclusion criteria and not meeting any of the exclusion criteria are randomized into one of the two treatment arms: ① standard therapy and TZV or ② standard therapy and TZV placebo. At each study site, patients are assigned to the trial group or placebo group according to the corresponding random numbers extracted at the time of inclusion in the study. Each study site will enroll 24 patients for a total of 240 patients across the ten study sites. A random number table generated using the randomization method in Statistical Analysis System software version 9.4 (SAS9.4) (SAS Institute Inc., USA) is used to assign the participants in a 1:1 ratio, with 120 patients in each of the trial and placebo groups.
《2.7. Electronic data collection and data management》
2.7. Electronic data collection and data management
2.7.1. Data collection
Data are managed using an electronic data capture (EDC) system. Study protocol and case report form (CRF)-related materials are provided to the clinical data management center along with the study’s data statistics in order to create an electronic CRF. The occurrence of unexpected issues during the study will be recorded, and the data management center will be informed in a timely manner.
2.7.2. Data management
According to the data verification plan, the data manager will check the data input in the EDC system and will conduct a comprehensive inspection of the data for missing information, logic problems, and issues concerning inclusion and exclusion criteria. After a data query from the data manager is confirmed by the PIs, study assistants will modify the data and respond to the query, and the data manager will cancel the query. This process will be repeated until all data in the database are confirmed to be correct.
2.7.3. Data monitoring
A DSMB has been established to monitor the conducting and results of the trial. The DSMB consists of multidisciplinary Chinese experts, including public health professionals, clinical specialists, an epidemiologist, a lawyer, and community health workers. The DSMB members have regular conferences via WeChat to review the protocol and study progress and to make recommendations throughout the whole study. The primary objectives are to ensure the safety of the study subjects and the integrity of the study data. The DSMB is responsible for giving advice on issues including research design, data quality and analysis, research participant protection, and interim analysis for the study.
2.7.4. Sample size calculation
Given the limited information about TZV for COVID-19 and the urgent clinical requirements, we expect to enroll 120 patients in each arm, assuming that the withdrawal rate due to death and other unanticipated conditions is less than 20%. The DSMB recommends that interim analysis be performed to monitor the safety of this trial and the effectiveness of the drug, which will help to adjust the sample size and decrease the risk of uncertainty during the trial implementation. The O’Brien–Fleming approximation of the Lan– DeMets spending function is used in our study to control type Ⅰ error.
2.7.5. Statistical analysis
The statistical analysis will be performed using SAS9.4, and the analysis will use an intention-to-treat approach to examine the differences between the two arms. Categorical variables will be described as counts and percentages, and continuous variables will be expressed as medians, interquartile ranges or means, and standard deviations, as appropriate. In the demographic analysis comparing the two groups, Pearson’s χ2 tests and Fisher’s exact tests will be performed for categorical variables. Other means for continuous data will also be compared using the t-test or the Mann– Whitney U test. In the analysis of outcomes efficacy, the Kaplan– Meier estimation will be used to estimate the overall survival curve of patients in different groups, and the differences in survival curves will be assessed using the log-rank test. The hazard ratio with respective 95% confidence intervals (CIs) was calculated by the Cox proportional hazards model. A P value lower than 0.05 will be considered to indicate statistical significance. A safety evaluation will be performed in all patients who have received at least one dose of TZV, and the rates of AEs and SAEs will be compared between the two groups. Exploratory subgroup analysis will be performed on COVID-19 patients with basic diseases such as diabetes and hypertension to consider the effects of these basic diseases on the efficacy and safety of the drug treatment.
《2.8. Outcome and measures》
2.8. Outcome and measures
2.8.1. Primary outcome
The primary outcome is the time to clinical improvement, which is the number of days from randomization to the return to normalization of the relevant symptoms, including body temperature, respiratory rate, fingertip oxygen saturation, alleviation of cough, and obvious absorption of pulmonary inflammation on chest CT images, along with maintenance of these results for at least 72 h. Normal body temperature is defined as an axillary temperature lower than 37.0 °C. The definition of normal respiratory rate is an indoor respiratory rate of fewer than 24 breaths per minute. Normal fingertip oxygen saturation is defined as an indoor oxygen saturation of greater than 94%. Alleviation of cough is defined as the reduction of the severity of cough to mild or absent using a physician-reported scale of severe, moderate, mild, or absent. Obvious inflammation absorption on chest CT images is defined as an absorption area with more than two-thirds of the area classified as lesions. The digital imaging and communications in medicine (DICOM) data of the chest CT images from each study site will be uploaded to the imaging center to ensure unified evaluations by the same group of experts.
2.8.2. Secondary outcomes
(1) Clinical improvement rate: the number of patients with clinical improvement among all of the intention-to-treat participants;
(2) Time for alleviation of fever: Alleviation of fever is defined as an axillary temperature lower than 37.0 °C that lasts for more than 24 or 72 h;
(3) Mean time and proportion of obvious inflammatory absorption in the lung;
(4) Conversion rate of repeated negative viral nucleic acid tests;
(5) Mortality on Day 28;
(6) Conversion rate of severe and critically severe patients. This outcome will be assessed on a six-point scale (Table 3). The subject being discharged or the score decreasing by two points is defined as a conversion from severe or critically severe to moderate, mild, or recovery.
《Table 3》
Table 3 Six-point scale of severe and critically severe patients.

2.8.3. Exploratory outcomes
The exploratory outcomes to be measured include routine blood and urine tests, coagulation function, and changes in inflammation indicators, including C-reactive protein and procalcitonin.
2.8.4. Safety outcomes
AEs, SAEs, liver function, kidney function, and concurrent treatments are monitored and recorded throughout the trial. Every study team was trained before the site initiation visit about the definitions of AEs. Every AE occurrence during the study—whether related to the study drug or not—and every occurrence during the follow-up period will be reported and recorded. The following AErelated information will be recorded: time of occurrence, severity, duration, actions taken, and outcome. In the case of SAEs, the events will be reported to the DSMB and the ethics committees of the local hospital within 24 h.
《3. Discussion》
3. Discussion
To date, no drugs have been proven to be successful and effective for the treatment of life-threatening COVID-19 caused by SARS-CoV-2 infection. Current treatment strategies are essentially empirical or tentative therapies and intensive care/life support, such as quarantine and isolation, early diagnosis, control of the infection source, personal protection to reduce transmission, meticulous supportive care, and supportive treatments for affected patients [5]. An RCT on remdesivir, a broad-spectrum antiviral nucleotide prodrug that was previously used to treat Middle East respiratory syndrome coronavirus (MERS-CoV) and severe acute respiratory syndrome coronavirus (SARS-CoV) infections [17,18], is ongoing in China with a sample size of 761, due to the effectiveness and safety profile of remdesivir on COVID-19. A series of clinical trials and cohort studies have been and are currently being conducted in Wuhan and other cities in China. More than 300 interventional or observational studies about COVID-19 are registered on the Chinese Clinical Trial Registry↑ or clinicaltrials.gov‡ , only ten of which are RCTs. The trial proposed herein is expected to provide first-hand evidence on the efficacy and safety issues of TZV for the treatment of COVID-19.
↑ https://clinicaltrials.gov/.
‡ http://www.chictr.org.cn/.
One strength of this trial is that it is an add-on interventional study consisting of the Chinese standard therapy plus the study drug or placebo. This design ensures protocol compliance and maximizes the facility of patient enrollment to this trial while maintaining the standard treatments according to the guidelines of the National Health Commission of the People’s Republic of China. Another strength is its block randomization in each of the ten study sites, in which the patients’ baseline demographic features and treatment characteristics will be recorded and balanced by randomization within each local hospital during the lifesupport treatment of patients in case of emergency.
There are some limitations to our study. First, all assessments of clinical symptoms and signs are conducted at local study sites by PIs and assistants without sufficient training on COVID-19 beforehand. Second, there is no central laboratory for urgent tests for diagnosing or monitoring disease with uniform sampletransferring procedures. Third, COVID-19 is a time-limited disease, and at present there are 100 confirmed COVID-19 patients in Heilongjiang Province. There is no guarantee that we will be able to acquire the prospective number of patients for the trial.
《4. Conclusions》
4. Conclusions
We designed this randomized clinical trial to investigate the efficacy and safety of TZV for COVID-19. Although the results of this study lack statistical significance due to the limited sample size, they are expected to show that TZV might benefit COVID-19 patients by controlling symptoms and reducing frequent usage of concomitant therapies for vital organ supports. There is a need for further research to be performed with large sample sizes to assess these outcomes.
《Acknowledgements》
Acknowledgements
We are deeply grateful to the front-line clinicians who participated in the study while directly fighting the epidemic. This study was supported by the Chinese Academy of Engineering Project for COVID-19 (2020-KYGG-01-04).
《Compliance with ethics guidelines》
Compliance with ethics guidelines
Xiaoke Wu, Kaijiang Yu, Yongchen Wang, Wanhai Xu, Hongli Ma, Yan Hou, Yue Li, Benzhi Cai, Liying Zhu, Min Zhang, Xiaoli Hu, Jingshu Gao, Yu Wang, Huichao Qin, Mingyan Zhao, Yong Zhang, Kang Li, Zhimin Du, and Baofeng Yang declare that they have no conflict of interest or financial conflicts to disclose.











 京公网安备 11010502051620号
京公网安备 11010502051620号




