
《1. Introduction》
1. Introduction
The coronavirus disease 2019 (COVID-19) pandemic, which is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), is quickly spreading throughout the world [1]. As of 26 June 2020, despite the rigid restrictions adopted by many countries worldwide, over 9.47 million total confirmed cases of COVID-19 have been reported in 188 countries and territories, resulting in approximately 0.48 million deaths. Since there are no effective drugs or vaccines for COVID-19 as yet, the rapid diagnosis and isolation of patients infected with SARS-CoV-2 have been regarded as the most effective way to restrain the ongoing pandemic. Unfortunately, COVID-19 shares many common symptoms with the flu and cold, such as fever, dry cough, and myalgia or fatigue [2]. As a result, reliable diagnostic tools for differentiating the novel coronavirus from other common respiratory viruses are greatly desired in the fight against COVID-19. So far, a variety of nucleic acid testing kits based on reverse transcription polymerase chain reaction (RT-PCR) have been developed and are playing a central role in clinically diagnosing COVID-19 [3,4]. However, these RT-PCR kits are usually time-consuming, can only detect SARSCoV-2, and occasionally produce false-negative results due to either sampling or sensitivity issues. In clinical practice, SARSCoV-2-negative patients tested by RT-PCR with persistent COVID19-like symptoms can still consume a great deal of precious medical resources and have a high risk of cross-infection if treated inappropriately. Therefore, it is extremely urgent to develop a diagnostic tool that can not only rapidly detect SARS-CoV-2, but also accurately identify other common respiratory viruses, such as influenza viruses, human parainfluenza virus (HPIV), and so forth [5–7], so that the exact cause of the symptoms can be known when the SARS-CoV-2 test is negative.
Multiplex RT-PCR is capable of detecting several types of viruses by mixing all the primers in a single tube. However, due to the inevitable interferences of these mixed primers and the limited number of available fluorophores for detection, the development of a robust multiplex system is extremely difficult and the number of detected targets is generally limited to less than ten per reaction [8]. One of the most promising methods for breaking this limitation is to transform a multiplex polymerase chain reaction (PCR) into a multiple single-plex PCR by physically isolating different primer pairs into designated compartments. In the past decades, microfluidic technology has become one of the most common methods to realize the sample partitioning strategy without introducing additional manual operations. Among various microfluidic technologies, such as hydrophilic–hydrophobic patterning [9,10], the SlipChip [11–14], and electrowetting-on-dielectric (EWOD) technology [15,16], the centrifugal-based method offers the advantages of simple operations, fully enclosed microstructures, and easy integration with other microfluidic functions [17,18]. Our group previously developed a disc-shaped centrifugal microdevice operated in a compact instrument with a single channel (only one sample tested per run) and proved the excellent multiplexing capability of this system by demonstrating the detection of 13 respiratory pathogenic bacteria using loop-mediated isothermal amplification (LAMP) on a single chip [19]. However, when viruses are detected, neither RT-PCR nor LAMP are amenable for use with a microchip due to thermal cycling and primer design problems, respectively. Several other microfluidic systems, including FilmArray® and Verigene®, have also been invented for multiplex pathogen detection and have the advantages of integration with sample preparations [20–22]. Unfortunately, no microfluidic systems have been reported as yet for distinguishing SARS-CoV-2 from other respiratory viruses.
Nucleic acid sequence-based amplification (NASBA) is an enzyme-based isothermal amplification method that employs a mixture of reverse transcriptase, ribonuclease (RNase) H, T7 ribonucleic acid (RNA) polymerase, two specially designed primers, and a specially designed probe [23,24]. In the initialreaction phase of NASBA, primer 1 anneals to the single-stranded RNA, synthesizes the complementary deoxyribonucleic acid (cDNA), and forms an RNA:DNA hybrid. Subsequently, RNase H hydrolyzes the RNA chain and generates single-stranded DNA. After annealing with primer 2, reverse transcriptase synthesizes double-stranded DNA with a promoter region that can be recognized by T7 RNA polymerase [23,24]. Once the double-stranded DNA with a promoter region is generated, the reaction enters into continuous cycles of transcription, reverse transcription, and hydrolysis of the RNA in the RNA:DNA hybrid [23–25]. Since 10– 1000 copies of RNA can be generated in each step of transcription, the NASBA process requires fewer cycles than PCR or LAMP, which reduces the total incubation time and overall error frequency [23,26]. As an isothermal amplification method, NASBA is robust, specific, and particularly suitable for the detection of singlestranded RNA, although it is not fit for double-stranded DNA [23,27].
In the current study, we demonstrate the development of a high-throughput, multi-index nucleic acid isothermal amplification analyzer (RTisochipTM-W) based on our disc-shaped centrifugal microfluidic technology with two major improvements. First, we chose NASBA, which is an efficient and sensitive isothermal RNA amplification technique [23,26,28,29], to replace the incompatible PCR and LAMP for detecting viruses in the centrifugal microchip. Second, we designed an array of 16 single-channel sub-modules, which were updated from the previous single-channel instrument, to increase the throughput of the system. As a result, this brandnew RTisochipTM-W system is capable of detecting 19 common respiratory viruses, including SARS-CoV-2, from 16 clinical samples in a single run within 90 min. The excellent performance of our system, which is proved by the thorough characterization in the current study, prompts us to envision this RTisochipTM-W system as playing a critical role in fighting the COVID-19 pandemic.
《2. Materials and methods》
2. Materials and methods
《2.1. Sample collections and RNA extractions》
2.1. Sample collections and RNA extractions
Plasmids containing the targeted virus sequences were synthesized by Sangon Biotech (Shanghai) Co., Ltd. RNA templates were prepared by in vitro transcription using a HiScribeTM T7 HighYield RNA Synthesis Kit (New England BioLabs, USA) according to the manufacturer’s instructions. All the inactivated virus culture stocks were given as a gift by Dr. Nan-Shan Zhong from the First Affiliated Hospital of Guangzhou Medical University. Clinical samples were collected with the patients’ informed consent from the Beijing Tsinghua Changgung Hospital, the Capital Institute of Pediatrics, the First Affiliated Hospital of Guangzhou Medical University, the West China Hospital of Sichuan University, and the Beijing Youan Hospital of Capital Medical University during the period of 2018–2020.
Throat swab samples were collected with a DNA flocked swab (93050, Miraclean Technology Co., Ltd., China) by rotating the swab tip in the throat to collect the cellular material; the swab was then placed into virus transport medium (MT0301, Youkang Hengye Biotechnology (Beijing) Co., Ltd., China). After the swab tip was immersed in the virus transport medium for a few seconds, the end of the swab tip was broken and the sample was maintained at –70 °C for long-term storage.
The total RNA from the clinical samples was extracted using TRIzolTM Reagent (Invitrogen, USA) or a QIAamp® Viral RNA Mini Kit (Qiagen, Germany) following the manufacturers’ instructions. Automated RNA extraction was performed with the KingFisherTM Flex Purification System (Thermo Fisher Scientific, USA) according to the user’s operation manual. The extracted RNA was dissolved in RNase-free water and quantified using a Nanodrop 2000 or Qubit 3.0 (all from Thermo Fisher Scientific, USA). The integrity of the RNA was examined using 1% formaldehyde denaturing gel electrophoresis.
《2.2. Primer design and NASBA reactions》
2.2. Primer design and NASBA reactions
The common respiratory viruses that can be detected by our system include SARS-CoV-2 (S and N genes), influenza A (H1N1, H1N1 2009, H3N2, H7N9), influenza B, respiratory syncytial virus (RSV), human metapneumovirus (HMPV), HPIV1, HPIV2, HPIV3, HPIV4, human rhinovirus (HRV), enterovirus 71 (EV71), coxsackievirus A16 (CA16), coxsackievirus A6 (CA6), human coronavirus (HCoV)-229E/NL63, and HCoV-OC43/HKU1. NASBA, an isothermal amplification technology, was employed for the on-chip detection of viral RNAs. The NASBA primers and probes were designed using the Primer Premier v.5.0 software (Premier Biosoft, USA) and synthesized by Sangon Biotech (shanghai) Co., Ltd., China. The sequences of primers and probes are listed in Table S1 of Appendix A. We also performed coverage analysis of the primers and probes of each pathogen; the results are listed in Supplementary data 2 of Appendix A. We downloaded ten recently published complete whole genome sequences of each pathogen to perform the coverage analysis. On the whole, the primers and probes designed in our system have very good coverage. For example, for H1N1 2019, the primers and probes totally matched with ten recently published complete whole genome sequences, indicating that the region we chose for primer and probe design is very conservative.
In the on-chip NASBA assay, a total of 1 μL of the home-made spotting solution containing each set of primers and probes was pipetted into each corresponding reaction chamber and dried completely at room temperature. During the analysis, a total of 55 μL of the total NASBA mixture containing 16.5 μL of the buffer solution, 13 μL of nucleoside triphosphate (NTP) solution, 5.5 μL of the enzyme mix (all from CapitalBio Technology, China), and 20 μL of RNA template were mixed in a tube and then injected into the chip with a pipette.
《2.3. Design and operation of the microfluidic chip》
2.3. Design and operation of the microfluidic chip
The microfluidic chip for the detection of 19 types of respiratory viruses has been reported previously by our group [30,31]. In brief, as illustrated in Fig. 1(a), the disc-shaped microfluidic chip has a diameter of 62 mm and a thickness of 0.6 mm; it is comprised of a structural layer and a thin cover layer connected by doublesided adhesive tape. The structural layer contains 24 reaction chambers (each 3 mm in diameter and 0.2 mm in depth) with a volume of 1.45 μL each, 24 buffer cells, a sine-shaped main channel (0.2 mm in width and 0.1 mm in depth), an inlet, and an outlet. Each reaction chamber is connected to the main channel via a buffer cell and a short pipe. The structural layer was fabricated of polycarbonate materials by precision injection molding, resulting in a smooth surface with a roughness of less than 10 μm. As shown in Fig. 1(b), each set of the NASBA primers and probes was pipetted into the designated reaction chamber and dried at room temperature. In addition, human genome DNA with a pair of primers targeting glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene was used as an internal positive control (IC). Plasmids of Saccharomyces cerevisiae together with the corresponding primers and probes were used as a positive control (PC) and sterilized nuclease-free water was employed as a negative control (NC). After the pre-fixing of primers, the structural layer and the cover layer were sealed together by the double-sided adhesive tape and pressed with a press machine. Each chip was individually vacuum-packed and stored at –20 °C before use.
《Fig. 1》

Fig. 1. Design of the disc-shaped microfluidic chip and workflow for the detection of respiratory viruses. (a) Schematic diagram of the microfluidic chip; (b) layout of the designated reaction chambers for each targeted virus and control on the chip; (c) mechanism of NASBA; in the non-cyclic phase, primer 1 anneals to the target sequence and forms an RNA:DNA hybrid. RNase H hydrolyzes the RNA from this RNA:DNA hybrid; next, primer 2 anneals to the single-stranded DNA (ssDNA) and synthesizes a doublestranded DNA (dsDNA) by reverse transcriptase (RT); in the cyclic phase, the reaction repeats the cycles of transcription of RNA from dsDNA templates, the synthesis of cDNA, the hydrolysis of the RNA chain in the RNA:DNA hybrid, and the formation of dsDNA; (d) workflow of virus detection. Left to right: receiving the inactivated clinical samples, extracting RNA from samples, loading reagents into the chip, sealing the chip with tape, loading the chip into the instrument, distributing reagents into the reaction chambers on the chip, and performing amplification and detection in the RTisochipTM-W system.
The mechanism of NASBA is shown in Fig. 1(c) and the entire analytical process of the RTisochipTM-W system is shown in Fig. 1(d). In brief, after the collection of a pharyngeal swab specimen, RNA was extracted from the clinical sample using a commercially available nucleic acid extraction kit and mixed with the reaction mixture of NASBA without primers and probes. Then, a total of 55 μL of the mixture was pipetted into the microchip through the inlet hole to fill the main channel. After that, the inlet and the outlet were sealed with two pieces of tape. Once the chip was loaded into the tray of the instrument and the start button in the control program was clicked, the rest of the procedure was carried out automatically by the system. The chip was first spun for two periods of time at a speed of 6000 revolutions per minute (rpm): first for 10 s, and then again for 30 s, after an interval of 120 s, leading to the loading of the reagents from the main channel into the 24 reaction chambers. The microfluidic chip was next incubated at 41 °C for 35 min. During this period, the fluorescence signals generated by the amplified products were detected and displayed on the computer screen in real time.
《2.4. Microfluidic instrument for the analysis of respiratory infectious diseases》
2.4. Microfluidic instrument for the analysis of respiratory infectious diseases
The RTisochipTM-W consists of four identical sub-modules, each of which can control and detect a microfluidic chip. Four analyzers can be piled up to form a 16-channel system that is controlled by a single computer (Fig. 2(a)). As shown in Figs. 2(b) and (c), the submodule is mainly composed of a motor, a control panel, a power module, a temperature-control module, a detection module, and a communication module. The control panel controls the rotation of the motor, the entry/exit of the microfluidic chip, and all the other modules. The proportional-integral-derivative (PID) temperature controller provides the temperature required for nucleic acid amplification. The optical detection system monitors the fluorescence signals in the reaction chambers in real time. Fluorescence data are sent simultaneously to the computer, which is connected with a router through the communication module.
《Fig. 2》
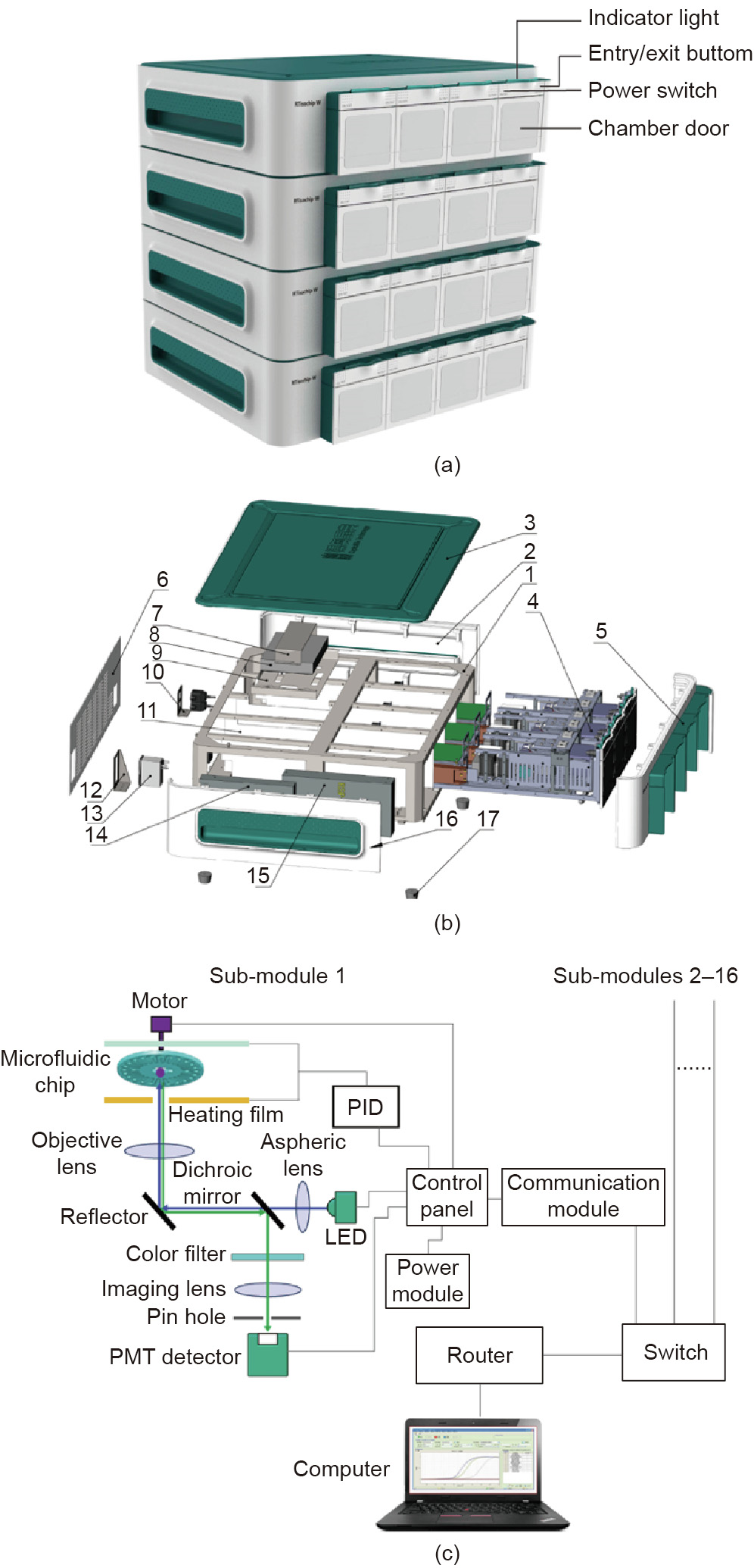
Fig. 2. Design of the RTisochipTM-W system. (a) Photograph of the 16-channel RTisochipTM-W with four piled analyzers, each of which contains four sub-modules. (b) Exploded view of the analyzer containing four sub-modules. 1: sheet metal frame; 2: right side panel; 3: upper panel; 4: sub-module; 5: front panel; 6: rear panel; 7: fixed upper cover panel of switch; 8: switch; 9: fixed panel of switch; 10: network port mounting panel; 11: partition panel; 12: power socket mounting panel; 13: socket-type power filter; 14: power supply for the system; 15: power supply for the microchip heater; 16: left side panel; 17: rubber foot. (c) Schematic of the structure of the sub-module inside the instrument. Up to 16 identical submodules can be connected to the computer via a router. PMT: photomultiplier tube; LED: light emitting diode; PID: proportional-integral-derivative.
《3. Results》
3. Results
《3.1. Optimization of RNA extractions for the RTisochipTM-W system》
3.1. Optimization of RNA extractions for the RTisochipTM-W system
The extraction of RNA from the clinical samples, as the first step of the analysis, plays a critical role in the entire analytical process. To select a suitable RNA extraction method, we compared the extraction efficiencies of the TRIzolTM universal total RNA extraction kit and the QIAamp® viral RNA mini kit for the extraction of viral RNA from pharyngeal swabs. The time to a positive value (Tp), defined as the time at the second derivative inflections of the exponential amplification curves, was employed as the critical factor to evaluate the extraction efficiencies, as Tp is negatively related to the template concentration in general. As shown in Fig. 3(a), we found that the Tp values provided by the TRIzolTM universal kit were always slightly shorter than those provided by the QIAamp® kit. Therefore, we chose the TRIzolTM kit as the recommended manual method for our system. Next, to further reduce the strength of manual operation, we employed the KingFisherTM Flex Purification System to automate the RNA extraction procedure. Figs. 3(b) and (c) demonstrate that this automatic system generally provided a higher extraction efficiency and better reproducibility than the manual TRIzolTM method in the detection of two viral targets, HCoV-229E/NL63 and H1N1 2009, in a wide range of virus concentrations represented by median tissue culture infectious dose (TCID50). However, due to the higher costs of the automated system, we chose both the TRIzolTM universal kit and the KingFisherTM system for the subsequent tests.
《Fig. 3》

Fig. 3. Comparison of different RNA extraction methods for on-chip virus detection. (a) Comparison of the RNA extraction efficiencies of the QIAamp® and the TRIzolTM kits by detecting four different virus targets (n = 3); (b) comparison of the manual TRIzolTM kit and the automated KingFisherTM Flex Purification System in the detection of HCoV-229E/ NL63 and H1N1 2009 (n = 3). All error bars represent one standard deviation. TCID50: median tissue culture infectious dose.
《3.2. Evaluation of the sensitivity and repeatability of the RTisochipTM-W》
3.2. Evaluation of the sensitivity and repeatability of the RTisochipTM-W
To evaluate the sensitivity of the platform, the synthesized RNA templates of these 19 respiratory viruses were diluted to form a concentration gradient from 250, 50, 25, to 10 copies·μL-1 . Among them, SARS-CoV-2 N and SARS-CoV-2 S were diluted together. The limits of detection (LODs) of all the viruses are listed in Fig. 4(a). We determined that the LODs of H7N9, CA16, RSV, HPIV1, HPIV2, HPIV3, HPIV4, HCoV-229E/NL63, and HMPV were 50 copies·μL-1 ; that those of H1N1, H3N2, influenza B, EV71, HCoV-OC43/HKU1, CA6, HRV, and SARS-CoV-2 N gene were 25 copies·μL-1 ; and that those of H1N1 2009 and SARS-CoV-2 S gene could even reach 10 copies·μL-1 . All these results are comparable to those of the conventional RT-PCR method. Typical amplification curves of H1N1 2009 and SARS-CoV-2 S at different RNA concentrations as well as the NCs are shown in Figs. 4(b) and (c). Good linear correlations were observed between the Tp values and the RNA concentrations in H1N1 2009 and SARS-CoV-2 S gene.
《Fig. 4》
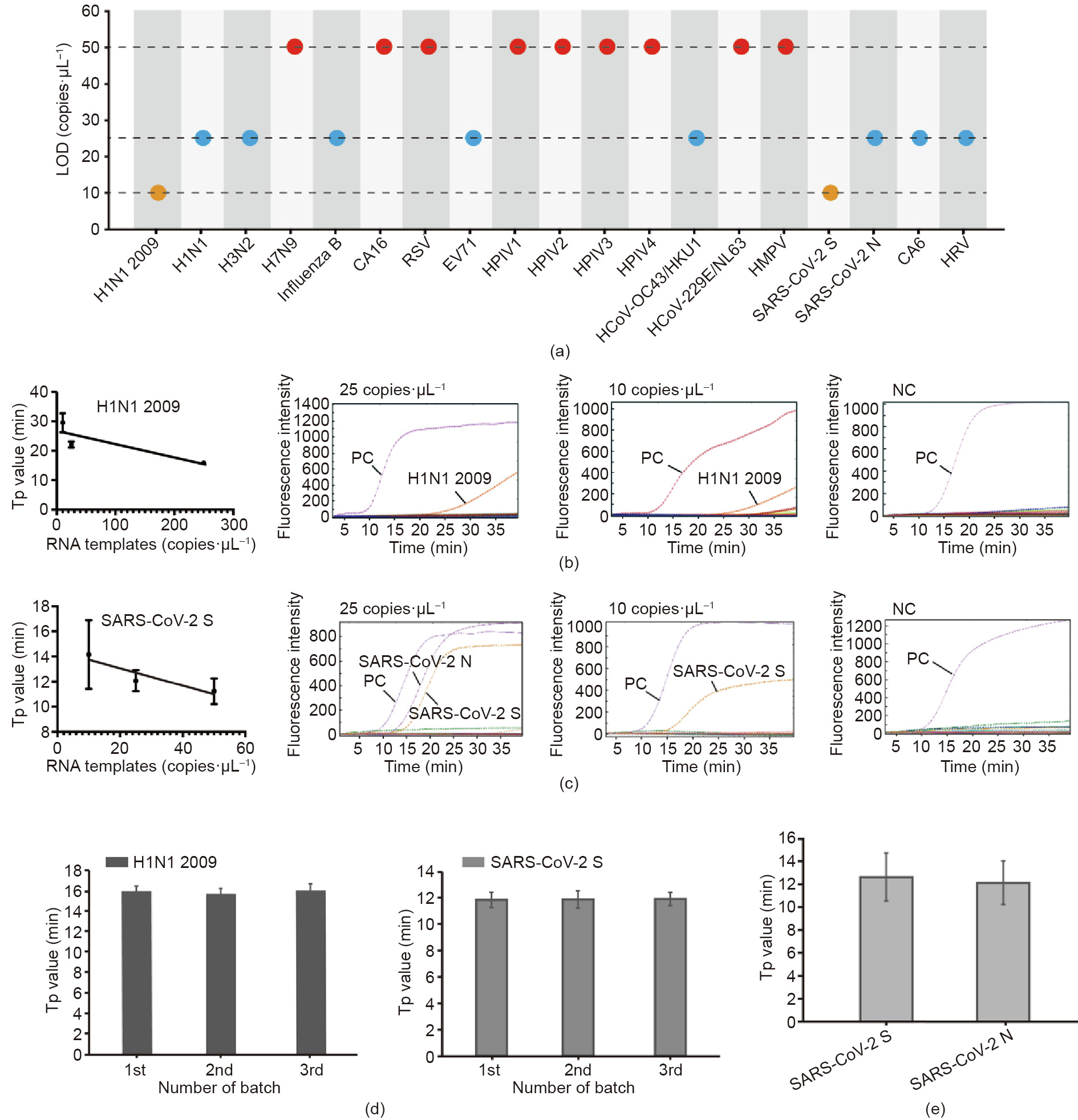
Fig. 4. Sensitivity and repeatability tests of the RTisochipTM-W system. (a) LOD of 19 respiratory viruses tested by the on-chip assays; (b) typical amplification curves and linear fitting between the Tp and the template concentration of H1N1 2009 (n = 3); (c) typical amplification curves and linear fitting of SARS-CoV-2 S gene (n = 3); (d) repeatability tests of three microfluidic chips with synthesized RNA templates of H1N1 2009 and SARS-CoV-2 S gene (n = 10); (e) repeatability of the detection of SARSCoV-2 S and N genes with extracted RNA samples (n = 10). All error bars represent one standard deviation.
We chose H1N1 2009 and SARS-CoV-2 as the detection targets for the evaluation of the system repeatability. First, three batches of the microfluidic chips (ten replicates in each batch) were tested with the inactivated virus stocks of H1N1 2009 and SARS-CoV-2 S gene at a concentration of 500 copies·μL-1 . After the RNA extraction, the prepared samples were loaded into the RTisochipTM-W system for testing. As shown in Fig. 4(d), no significant differences were observed in terms of the Tp values of these two targets within each batch and among three batches, indicating the excellent repeatability of our RTisochipTM-W system. After the evaluation with mock samples, we further selected SARS-CoV-2 S and N genes as targets to test the repeatability with RNA extracted from clinical samples. Fig. 4(e) shows that the coefficients of variation (CVs) of the S and N genes with ten repeats were 16.96% and 15.91%, respectively. These results clearly demonstrate that our microfluidic platform can provide acceptable sensitivities with high repeatability for multi-index virus detection.
《3.3. Evaluation of the robustness and specificity of the RTisochipTM-W system》
3.3. Evaluation of the robustness and specificity of the RTisochipTM-W system
Robustness is a crucial feature of a medical diagnostic platform, especially for the diagnosis of infectious diseases, because clinical samples may come in various forms and contain diverse amplification inhibitors that cannot be eliminated by the extraction process. In our study, the pharyngeal swabs or other types of specimens could have had multiple interferents, including microorganisms, blood, and residual antibacterial and antiviral drugs that the patients took prior to sampling. Thus, we thoroughly evaluated the anti-interference capability of our platform using samples with artificially added interferents. First, as shown in Fig. 5(a), we found that that the addition of mucin at a final concentration of 10 mg·L-1 into a 1 × 104 copies·mL-1 virus mixture of H1N1 2009, H3N2, influenza B, RSV, HPIV3, and SARS-CoV-2 N had no negative impacts on the amplification of these five targets, as the Tp values were not changed significantly. Next, we prepared a mixture of interfering substances, including oxymetazoline hydrochloride, dexamethasone, interferon, lamivudine, amantadine, menthol, sodium chloride, and mucoprotein (listed in Fig. 5(b)), in order to more critically evaluate the anti-interference performance of the platform. As shown in Fig. 5(c), the Tp values of H1N1 2009, H3N2, influenza B, RSV, HPIV3, and SARS-CoV-2 N gene showed no significant changes after these mixed interferents were added. Nevertheless, these tests proved that the interferents presented in the samples had no significant negative effects on our RTisochipTM-W platform.
Since our platform can detect 19 different types of viruses simultaneously, the cross-reactions among these targets need to be thoroughly investigated, as a cross-reaction may induce falsepositive results in the tests. Here, we prepared 19 samples, each of which contained a single type of virus template (P1–P19), and six NCs (N1–N6) to test our system. As shown in the heatmap of Fig. 5(d), we found that there was no non-specific amplification caused by the cross-reactions in all of the 25 samples, indicating the reliability of the RTisochipTM-W system. Next, we noticed that the competitive effect within these 19 different targets is another potential risk for the platform. When multiple targets are present in a single sample, our system might be less sensitive than a system that is detecting only one target at a time, due to interference from the non-targeted RNA background. To evaluate this competitive effect, we prepared single- or mixed-RNA samples that were diluted to final concentrations of 5000 and 500 copies·μL-1 . As shown in Fig. 5(e), the Tp values of H1N1 2009, H3N2, RSV, HPIV3, SARS-CoV-2 S, and SARS-CoV-2 N detected individually were not significantly different from those obtained from the mixed RNA samples at the concentrations of 5000 and 500 copies·μL-1 . Although we did observe that influenza B was not detectable in the mixed samples at the lower concentration due to the non-targeted RNA background, the primer concentrations and primer sequences of influenza B can be further optimized to resolve this problem. Overall, the above results proved that the RTisochipTM-W system possesses good specificity, strong robustness, and a powerful testing capacity for detecting 19 respiratory viruses from 16 clinical samples at the same time.
《Fig. 5》

Fig. 5. Anti-interference, cross-reaction, and competitive effect tests of the RTisochipTM-W. (a) Similar Tp values and amplification curves of seven selected targets in reactions with or without mucin. (b) Final concentrations of the interfering substances added to the reaction buffer. (c) Similar Tp values of seven selected targets in reactions with or without mixed interfering substances. (d) Cross-reaction tests of the platform. Positive results are shown as orange squares, while azure squares represent negative results. P1–P19: virus templates; N1–N6: NCs. (e) Competitive effect tests of mixed target samples. Two different concentrations of single- or mixed-target samples were tested and influenza B was missed at the low concentration of 500 copies·μL-1 . ns: not significant.
《3.4. Clinical tests of SARS-CoV-2 and another 18 respiratory viruses using the RTisochipTM-W》
3.4. Clinical tests of SARS-CoV-2 and another 18 respiratory viruses using the RTisochipTM-W
Prior to the outbreak of SARS-CoV-2, we had followed the epidemic trends of 18 types of respiratory viruses in Beijing by analyzing a large number of clinical samples using the RTisochipTM-W system. During the flu season from 27 December 2019 to 16 January 2020, a total of 101 flu-season samples of children’s throat swabs were collected by the Beijing Tsinghua Changgung Hospital. As a comparison, 100 non-flu-season samples of children’s throat swabs were collected by the Capital Institute of Pediatrics between 18 September and 7 November 2019. All clinical samples were tested by the RTisochipTM-W system; the results are listed in Tables S2 and S3 of Appendix A. From these 201 clinical samples, we found that the positive rates of these respiratory viruses showed large variations over the sampling time. As shown in Fig. 6(a), almost 71.3% of the flu-season samples were found to be infected with influenza viruses, in comparison with only 2.9% of the nonflu-season samples from Capital Institute of Pediatrics. Furthermore, the positive ratios of RSV, HPIV1, HPIV2, HMPV, HCoV229E/NL63, and HCoV-OC43/HKU1 were on the rise in the flu season. Conversely, the positive ratios of HRV, HPIV3, and HPIV4 were lower in the flu-season samples than in the non-flu-season samples. The significant changes in the positive rates of respiratory viruses over time underlined the importance of the multi-index detection of respiratory viruses in clinics.
After the outbreak of SARS-CoV-2, we added SARS-CoV-2 S and N genes to the system and evaluated the system performance using clinical SARS-CoV-2 samples. First, 14 clinically confirmed positive and three negative samples were tested by the RTisochipTM-W system and the commercial RT-PCR kit (BGI, China) in parallel. As summarized in Fig. 6(b) and Table S4 of Appendix A, both methods showed a 100% consistency, indicating that the detection performance of our platform was the same as that of conventional RTPCR. To further verify the stability and repeatability of our platform for testing clinical samples, we prepared a total of 25 microfluidic chips manufactured in three different batches to test 25 clinically confirmed samples. As illustrated in the heat map in Fig. 6(c), the results reported by our RTisochipTM-W system were all consistent with the clinical records, and no false-positive or false-negative results were obtained. After these small-scale evaluations, we collected a total of 614 clinical swab samples from suspected or confirmed COVID-19 patients in collaboration with the First Affiliated Hospital of Guangzhou Medical University (380 cases), the West China Hospital of Sichuan University (63 cases), and the Beijing Youan Hospital of Capital Medical University (171 cases) (listed in Tables S5–S8 of Appendix A). As shown in Fig. 6(d), the positive rates of SARS-CoV-2, H1N1 2009, H3N2, HPIV, RSV, and influenza B reported by our system were 6.19% (38/614), 10.91% (67/614), 6.19% (38/614), 3.75% (23/614), 12.38% (76/614), and 5.21% (32/614), respectively. The total coincidence rates of the RTisochipTM-W with the referenced kits (RT-PCR, Shanghai ZJ BioTech Co., Ltd., China) in the tests of SARS-CoV-2, H1N1 2009, H3N2, HPIV, RSV, and influenza B were 98.15%, 98.70%, 99.35%, 99.57%, 97.61%, and 99.35%, respectively. In these clinical samples, some suspected COVID-19 patients were eventually diagnosed as influenza A, influenza B, or RSV instead of SARS-CoV-2. For example, case No. 1 from the West China Hospital of Sichuan University was diagnosed as influenza A, highlighting the value of our platform for precisely identifying the exact cause of diseases. In some clinically confirmed COVID-19 cases, neither our platform nor conventional RT-PCR methods could detect SARS-CoV-2, probably due to the low viral loads of the samples. Such a large scale of clinical tests clearly proved that our platform is a powerful and reliable diagnostic tool in the detection of respiratory viruses in clinical settings.
《Fig. 6》
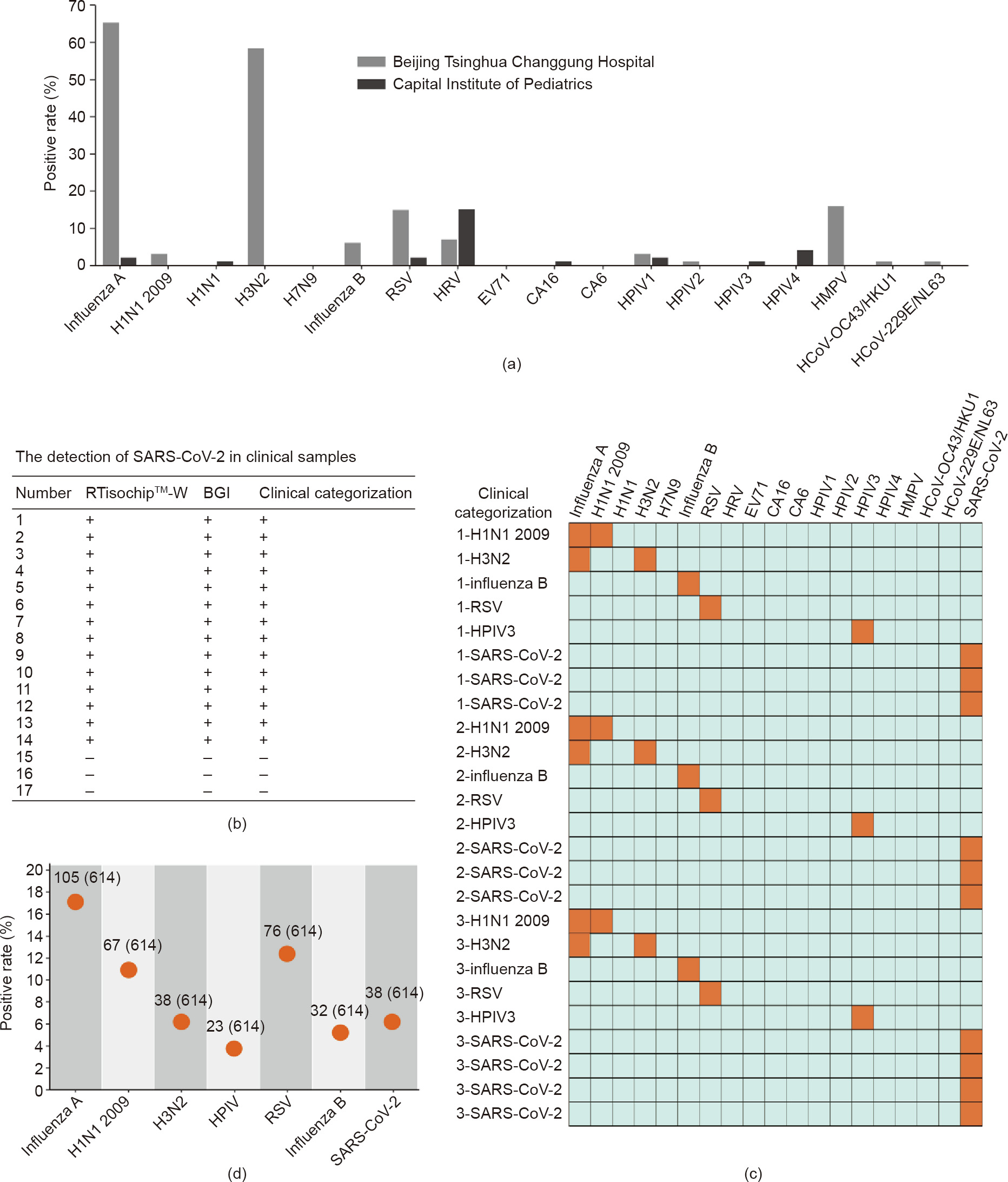
Fig. 6. Clinical identification of respiratory viruses using the RTisochipTM-W. (a) Positive rates of each virus target in clinical samples collected in 2019–2020. Samples from the Beijing Tsinghua Changgung Hospital were collected in the flu season from 2019 to 2020 (n = 101), and samples from the Capital Institute of Pediatrics were collected in the non-flu season of 2019 (n = 100). (b) Comparison of the SARS-CoV-2 detection results obtained by the RTisochipTM-W and the conventional RT-PCR. "+” represents a positive result and "–” is negative. (c) Repeatability tests of the RTisochipTM-W using clinical samples. Positive and negative results are shown as orange and azure squares, respectively. (d) Positive rates of a total of 614 clinical throat swab samples from suspected or confirmed COVID-19 patients. The detailed number was indicated.
《4. Discussion》
4. Discussion
Since there is no effective drug for COVID-19 at present, the only feasible way to fight against COVID-19 is to diagnose, classify, and isolate patients as early as possible. Unfortunately, due to the similar symptoms of all respiratory infection diseases, it is not uncommon to find that a SARS-CoV-2-negative patient with the symptoms of cough and fever has been admitted to an intensive care unit (ICU) room with other COVID-19 patients and has been treated as having COVID-19 in a medical resource-constrained situation. From this perspective, the development of a nucleic acid testing tool for distinguishing COVID-19 from cases of pneumonia caused by other viruses—such as influenza viruses, parainfluenza virus, adenovirus, RSV, rhinovirus, and HMPV—is extremely valuable. The RTisochipTM-W system is capable of identifying 19 common respiratory viruses in a single test and possesses the advantages of a short turnaround time of less than 90 min, a flexible throughput from one to 16 samples, minimum manual operations for multi-index detections, a sensitivity that is comparable to that of conventional RT-PCR, and a high tolerance for various interferents. Therefore, the RTisochipTM-W system should be able to play a significant role in fighting the COVID-19 pandemic.
Application of the RTisochipTM-W system in clinical practice should be elaborately planned within the context of the pandemic. Here, we present a guideline of testing for COVID-19 by comprehensively considering the pros and cons of all the molecular diagnosis methods. As shown in Fig. 7, first, when patients with COVID19-like symptoms arrive at the fever outpatient department, a conventional RT-PCR test can be administrated to rapidly identify whether the patients (and their close contacts) have been infected with SARS-CoV-2. Due to its relatively low cost and high throughput, assisted by robotics, RT-PCR is suitable for screening a large number of samples in a pandemic situation. If a patient is diagnosed as COVID-19, he or she could be moved to a designated COVID-19 hospital. During treatment, the viral loads of the patients can be closely monitored using digital PCR (dPCR), which can provide an absolute virus quantitation. Recent studies have shown that the viral load within the patient’s body reflects the treatment outcomes [32–34]. If the patient’s status deteriorates, the RTisochipTM-W system can be utilized to identify the presence of any hospital-acquired infections (HAIs) caused by respiratory viruses and bacteria. This testing strategy should be able to improve the survival rate of COVID-19 patients. Second, if patients at the fever outpatient department test as negative in the SARSCoV-2 tests, our RTisochipTM-W could soon identify whether the symptoms are caused by other known viruses and bacteria in the pathogen list of the system. Once diagnosed, the patients could be moved to the pneumology department of the hospital and be closely monitored by the RTisochipTM-W. Third, if the patients at the fever outpatient department still test as negative, nextgeneration DNA/RNA sequencing could be used to identify any unknown or uncommon pathogens. Then, the patients could be treated properly in the pneumology department and be monitored by our RTisochipTM-W for any sign of HAI. Finally, the recovered patients should accept another round of RT-PCR tests before being discharged. Meanwhile, considering the possible false-negative results of RT-PCR, immunoglobin M (IgM)- and immunoglobin G (IgG)-based immunoassays should be coupled with RT-PCR to improve the accuracy of the test, as recommended by the Diagnosis and treatment protocol for novel coronavirus pneumonia (trial version 7) [35].
《Fig. 7》
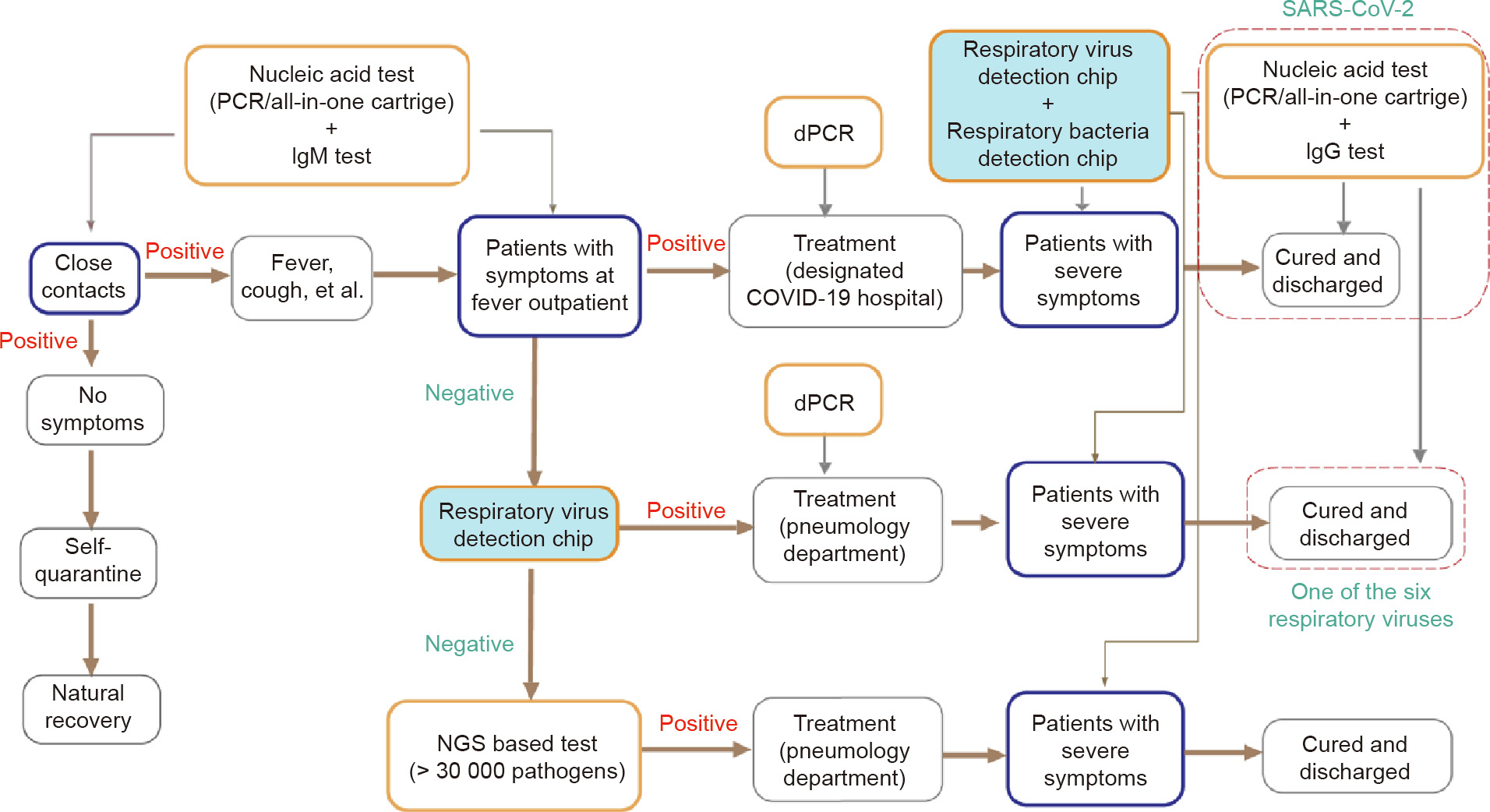
Fig. 7. COVID-19 testing guideline. NGS: next generation sequencing; dPCR: digital PCR; IgM: immunoglobin M; IgG: immunoglobin G.
Based on this guideline, the RTisochipTM-W system can be used in the following scenarios: ① to rapidly diagnose the exact cause of COVID-19-like symptoms in most cases in order to prevent mistreatment and cross-infection among patients; and ② to closely monitor the occurrence of HAIs during treatment due to the deterioration of medical situations caused by the pandemic. The RTisochipTM-W system, synergistically combined with other in vitro diagnostic tools, will make our fight against COVID-19 more efficient and more precise. In the future, the capability of the RTisochipTM-W can be further improved by introducing more pathogens into the detection list, by integrating sample preparations to form a ‘‘sample-in-answer-out” system, and by combining with an automated robotic system to reach higher throughput.
《 5. Conclusions》
5. Conclusions
In summary, we have successfully developed a high-throughput nucleic acid isothermal amplification system that is capable of detecting 19 common respiratory viruses, including SARS-CoV-2, from 16 clinical samples within 90 min. The high sensitivity, good specificity, strong robustness, and excellent repeatability demonstrated in the extensive clinical tests proved that our RTisochipTMW system can be utilized as a powerful nucleic acid testing tool in the diagnosis and screening of respiratory viruses, including SARS-CoV-2, in fever outpatient departments, ICUs, medical quarantine areas, and many other settings, where the precise cause of pneumonia is urgently demanded.
《Acknowledgements》
Acknowledgements
We thank Dr. Nan-Shan Zhong and his colleagues from the First Affiliated Hospital of Guangzhou Medical University for providing the inactivated virus culture stocks. We thank Dr. Xiuying Zhao and her colleagues from the Beijing Tsinghua Changgung Hospital and Dr. Xiaodai Cui and his colleagues from the Capital Institute of Pediatrics for providing clinical samples. This work is funded by the National Key Research and Development Program of China (2020YFC0847400) from the Ministry of Science Technology of China, the Project of Science and Technology Emergency Response for Prevention and Control of COVID-19 from the Tsinghua University (20201080055) and the Guangzhou Institute of Respiratory Health (2020GIRHHMS02 and 2020GIRHHMS03).
《Compliance with ethics guidelines》
Compliance with ethics guidelines
Wanli Xing, Yingying Liu, Huili Wang, Shanglin Li, Yongping Lin, Lei Chen, Yan Zhao, Shuang Chao, Xiaolan Huang, Shaolin Ge, Tao Deng, Tian Zhao, Baolian Li, Hanbo Wang, Lei Wang, Yunpeng Song, Ronghua Jin, Jianxing He, Xiuying Zhao, Peng Liu, Weimin Li, and Jing Cheng declare that they have no conflict of interest or financial conflicts to disclose.
《Appendix A. Supplementary data》
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.eng.2020.07.015.











 京公网安备 11010502051620号
京公网安备 11010502051620号




