

《1. Introduction》
1. Introduction
In 2020, coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has disrupted societies around the world. The five most common signs and symptoms of COVID-19 are fever, cough, fatigue, myalgia, and dyspnea [1]. While mortality rates are low in healthy people below 65 years, the elderly and those with comorbidity are far more prone to serious outcomes, including death [2,3].
Given the lack of effective antiviral therapy against COVID-19, current treatments mainly focus on symptom and respiratory support [4]. However, significant efforts have been put into identifying drugs for the treatment of COVID-19, and chloroquine, remdesivir, convalescent plasma, and immunoglobulin G transfusion have all been evaluated, although the effectiveness of any of these agents has not been proven [5,6].
Triazavirin (TZV), a new antiviral drug, has been on the market in Russia since 2015. The main principle of action of TZV is to inhibit the synthesis of viral ribonucleic acid (RNA) and the replication of viral genomic fragments through its synthetic analogue to the bases of purine nucleosides [7–9]. A phase II clinical trial of TZV showed that this compound significantly reduces the duration of the main clinical symptoms of influenza (intoxication, fever, and respiratory symptoms) and decreases the incidence of influenzarelated complications and the use of symptomatic drugs [10]. However, the efficacy of TZV for COVID-19 is unknown. We conducted a multicenter placebo-controlled trial on this subject.
《2. Methods》
2. Methods
《2.1. Study design》
2.1. Study design
We performed a multicenter, double-blinded, randomized controlled trial (RCT) in ten sites in Heilongjiang Province in Northern China. The study protocol was approved by the institutional review boards of all local sites, and all participants provided written informed consent. The study was chaired by a multidisciplinary steering committee. An independent data and safety monitoring board (DSMB) monitored the trial following standardized adverse-event reporting procedures. Independent study monitors attended all study procedures and verified all recorded data. This trial is registered on the Chinese Clinical Trial Registry with the identifier number ChiCTR20000300001 and the study protocol was recently published [11].
《2.2. Ethics》
2.2. Ethics
The clinical trial was carried out according to the principles of the Declaration of Helsinki and followed the laws, regulations, and administrative provisions of the Health Commission of Heilongjiang Province. The trial commenced after the approval of the Ethic Committees was obtained. Participants were informed of the risks and benefits of the study and were allowed to voluntarily cease participation in the study at any time for any reason. To protect the privacy of the subjects, each patient was identified with a unique random number, and patients’ names and personal information were kept confidential to everyone except for the researchers.
《2.3. Eligibility criteria》
2.3. Eligibility criteria
COVID-19 patients were recruited from the emergency departments, isolation wards, and intensive care unit (ICU) inpatient departments of the study sites. The inclusion criteria were: ① laboratory-confirmed SARS-CoV-2 infection by real-time reverse transcription polymerase chain reaction (RT-PCR); ② chest computed tomography (CT) imaging-confirmed lung damage—namely, multiple small plaques and stromal changes in the lungs, manifested in the outer lung, or with multiple ground glass shadows and infiltration shadows in both lungs (although these changes might not be present in mild patients); ③ hospitalized patients with fever (axillary temperature ≥ 37.0 °C) or respiratory symptoms; ④ a time from symptom onset to randomization of less than 12 d; ⑤ not having participated in other clinical research within the past three months; ⑥ 18 years or older; ⑦ not participating in other antiviral studies within 28 d of follow-up; and ⑧ written informed consent. Baseline laboratory, anthropometric, and clinical measurements, including chest CT imaging and throat swab test by laboratory RT-PCR, were performed at the local sites.
The exclusion criteria were: ① patients who were unsuitable or who could not participate safely in the study, as judged by the principle investigators; ② patients with serious grade C liver disease, according to the Child–Pugh score; ③ patients with severe renal impairment (glomerular filtration rate ≤30 mL· (min·1.73 m2 ) –1 ) or continuous renal replacement therapy, hemodialysis, or peritoneal dialysis; ④ patients with severe anemia (hemoglobin < 60 g·L–1 ); ⑤ pregnant or breastfeeding women; ⑥ patients with a history of allergy to TZV or its metabolic components; ⑦ patients with the possibility of being transferred to another hospital within 72 h of randomization; and ⑧ patients who participated in other clinical trials for COVID-19 within 30 d prior to our screening.
《2.4. Randomization and blinding》
2.4. Randomization and blinding
Randomization, stratified by study site, was based on a computer-generated allocation sequence; the 1:1 allocation sequence used permuted, random block sizes of four. The investigators who were responsible for assessing the primary outcomes and the patients were blinded to the study group assignment.
《2.5. Intervention》
2.5. Intervention
Participants at each site had their standard treatment supplemented with a daily oral dose of the trial drug TZV or its placebo. Participants with a mild or ordinary condition took 250 mg orally three times a day (at 9:00, 13:00, and 17:00), respectively, for seven consecutive days, while participants with a severe or critical condition took 250 mg orally four times a day (at 9:00, 13:00, 17:00, and 21:00), respectively, for seven consecutive days. A patient’s condition was considered to be mild/ordinary if symptoms such as fever and respiratory tract symptoms were present, and signs of pneumonia could be seen on imaging. A severe condition was defined as a respiratory rate ≥ 30 breaths per minute, pulse oxygen saturation (SpO2) ≤93% on room air at rest state, arterial partial pressure of oxygen (PaO2)/fraction of inspiration oxygen (FiO2) ≤300 mmHg (1 mmHg ≈ 133.322 Pa), or > 50% lesions progression within 24–48 h in pulmonary imaging. A critical condition was defined as respiratory failure resulting in mechanical ventilation, shock occurrence, or the occurrence of other organ failure requiring monitoring and treatment in the ICU.
Drugs were administered by qualified medical or nursing staff. The first dose of TZV was administered immediately after randomization. In addition to the study drug or placebo, all patients received routine standard therapy, according to the Chinese guidelines for COVID-19 [4,12].
The study drug TZV was manufactured by Zavod Medsintez, Ltd. (Novouralsk, Russia), and the placebo was manufactured by Tianqing Pharmaceutical (High-Tech Development Zone, Harbin, China). The drug and placebo needed for the study were repackaged in identical bottles with the same labels at a Chinese pharmaceutical factory. If a patient withdrew from the trial, the medication was stopped and recalled.
The medication period was 7 d, and the clinical symptoms, vital signs, fingertip oxygen saturation, and adverse events were recorded every day. Electrocardiograms, blood gas analysis, and other laboratory tests were performed on days 3 and 7, and CT was performed on day 7. The follow-up period was 28 d or until clinical improvement was observed, and the corresponding examinations were carried out at local hospitals on days 8, 9, 10, 14, 21, and 28 of the follow-up period.
《2.6. Sample size calculation》
2.6. Sample size calculation
Due to the limited information on TZV for COVID-19 and the urgent clinical requirements, we expected that enrolling 120 patients in both arms would result in a withdrawal rate due to death and other unanticipated conditions of less than 20%. After the recruitment of 52 participants, the DSMB recommended terminating the trial because no more daily new cases were added for a continuous week.
《2.7. Statistical analysis》
2.7. Statistical analysis
Categorical variables are described as counts and percentages, and continuous variables are expressed as median, interquartile range (IQR) values, mean, and standard deviation. Proportions for the categorical data were compared using the χ2 test or Fisher’s exact test. Means for continuous data were compared using independent group t-tests if the data were normally distributed; otherwise, the Mann–Whitney U test was used. No imputation was made for missing data.
A Cox proportional hazard ratio model was used to determine whether there were differences in the alleviation of symptoms. For unadjusted comparisons, a two-sided significant level (α) of less than 0.05 was considered statistically significant. The analyses were not adjusted for multiple comparisons. All statistical analyses were performed using the SAS software (version 9.4).
《2.8. Outcomes》
2.8. Outcomes
2.8.1. Primary outcome
The primary endpoint was time to clinical improvement, defined as the days from randomization until normalization. Clinical improvement was assessed by five components including body temperature, respiratory rate, oxygen saturation, alleviation of cough, and absorption of pulmonary infection by chest CT. Normalization was defined as body temperature < 37.0 °C, respiratory rate < 24 times per minute indoors, and oxygen saturation > 94% (fingertip). Alleviation of cough was defined as a reduced severity of cough from a physician-reported scale of severe or moderate to mild condition or absence. Absorption of pulmonary infection was defined as an absorption area > 2/3 by Digital Imaging and Communications in Medicine (DICOM) images on chest CT. Alleviation of these five clinical symptoms were required to stay normal for at least 72 h on all components to fulfill the primary endpoint of clinical improvement.
2.8.2. Secondary outcomes
Prespecified secondary outcomes included clinical improvement rate, median time and proportion of defervescence, mean time and proportion of significant inflammatory absorption of lung lesions, a negative conversion rate of the viral nucleic acid test, mortality at day 28, and the conversion from mild or ordinary to severe or critical severe status. The diagnosis and classification of mild, ordinary, severe, and critical severe conditions of this disease were set according to Chinese guidelines for COVID-19 [4,12].
Defervescence time was defined as the days from randomization to a temperature less than 37.0 °C maintained for at least 24 or 72 h. A throat swab viral nucleic acid test was repeated after 48 h following a negative test result. The conversion rate in severe and critical severe subjects was assessed with a six-point scale. The patient being discharged from the hospital or the score decreasing by two points from the baseline was considered to be conversion.
2.8.3. Exploratory outcomes
The exploratory outcomes were changes in laboratory indicators, including routine blood test, C-reactive protein (CRP), coagulation function, myocardial enzymes, and hepatic and renal functions. The recovery rates of the above indicators from abnormal to normal were also calculated.
《3. Results》
3. Results
During the study period, we screened 245 patients, of which 111 were eligible and 68 signed informed consent. A total of 52 patients with COVID-19 and laboratory-confirmed infection underwent randomization (Fig. 1). The first participant was enrolled on 14 February 2020, and the trial ended on 6 March 2020. The trial was terminated ahead of schedule by the decision of the DSMB because the COVID-19 outbreak in China was under control at the time, and there had been no new cases for one week.
《Fig. 1》
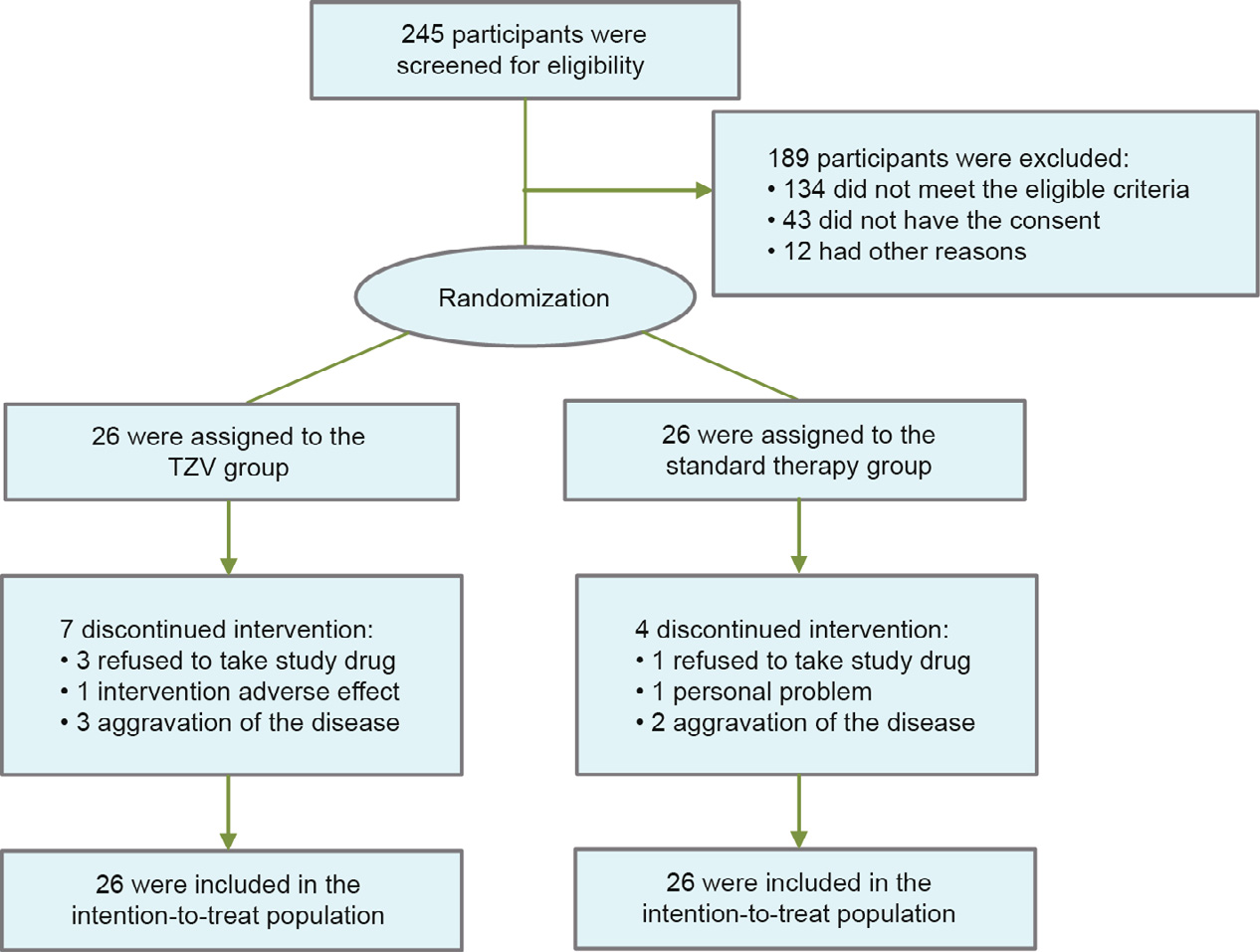
Fig. 1. Flow diagram of participants in the trial.
Among the 52 patients, 26 were assigned to the TZV group and 26 to the placebo group. The most frequent comorbidities were hypertension (28.8%), cardiovascular disease (15.4%), diabetes (15.4%), cerebrovascular disease (7.7%), and chronic obstructive pulmonary disease (COPD) (5.8%). Seven patients (26.9%) in the TZV group and four patients (15.4%) in the placebo group discontinued taking drugs varying from the second day to the sixth day of the study. The reasons for discontinued intervention are listed in Fig. 1. All participants were included in the intention-to-treat analysis.
Baseline characteristics are shown in Tables 1 and S1 of Appendix A. The median age of the patients was 58 years (IQR, 48– 65 years), and 50% were male. The median interval time between symptom onset and randomization was 7 d (IQR, 5–10 d). The most common four signs and symptoms were dry cough (55.8%), nasal obstruction (38.5%), pharyngalgia (32.7%), and fever (28.8%). Among the 52 patients, 44 (84.6%) had abnormalities on chest CT, 30 (57.7%) had bilateral patchy ground glass shadow, 18 (34.6%) had consolidation, and two had pleural effusion (3.9%).
《Table 1》
Table 1 Demographics and baseline clinical characteristics of the patients.
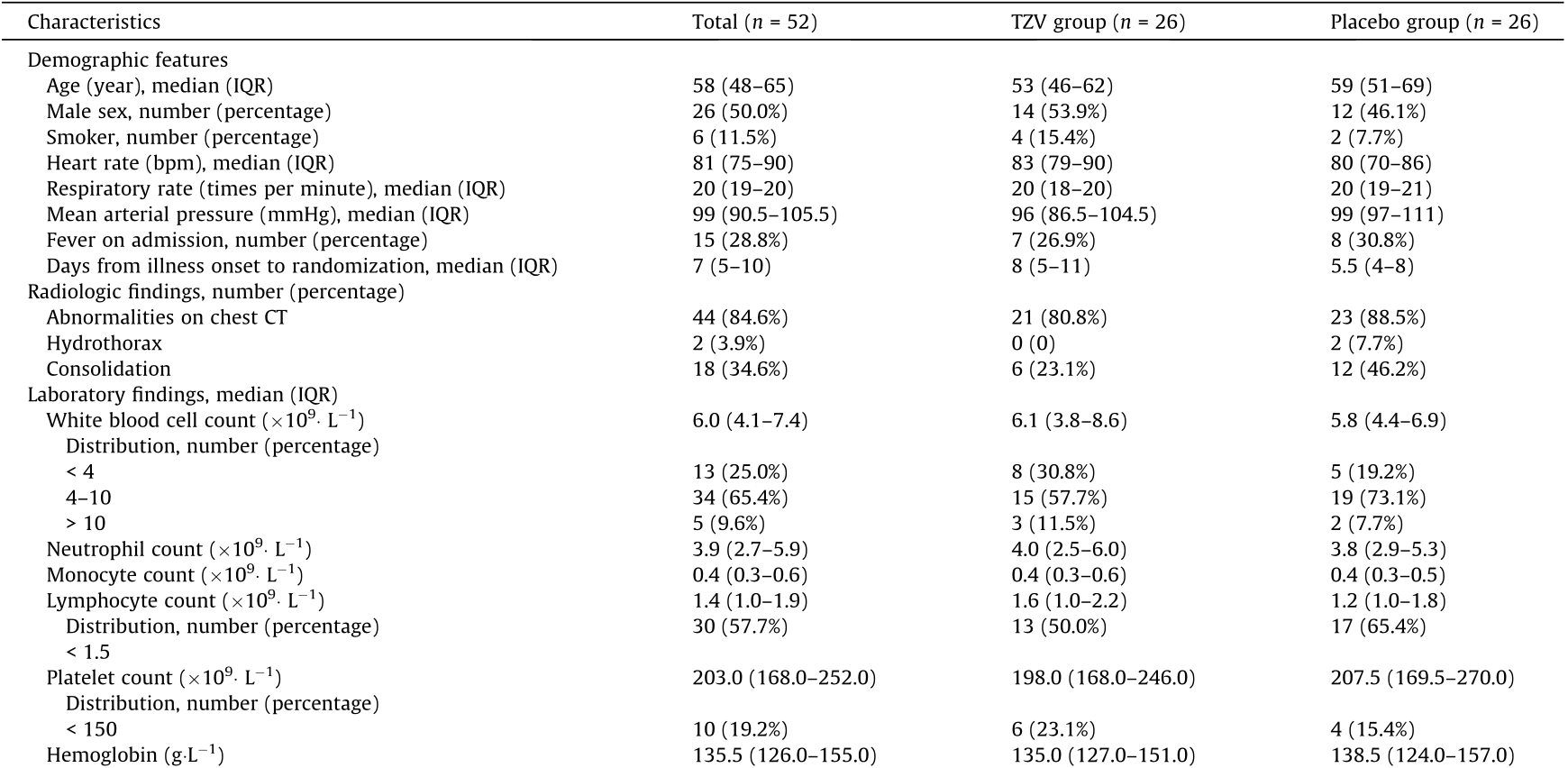

Lymphocytopenia is defined as a lymphocyte count of less than 1.5 per cubic liter. Thrombocytopenia is defined as a platelet count of less than 150 per cubic liter. bpm: beat per minute; PT: prothrombin time; APTT: activated partial thromboplastin time; LDH: lactate dehydrogenase; CK-MB: creatine kinase–MB; α-HBDH: α-hydroxybutyrate dehydrogenase; ALT: alanine aminotransferase; AST: aspartate aminotransferase.
《3.1. Primary outcome》
3.1. Primary outcome
The time to clinical improvement was 7 d after TZV versus 12 d after placebo (risk ratio (RR), 2.0; 95% confidence interval (CI), 0.7– 5.6; p = 0.2; Table 2).
《Table 2》
Table 2 Clinical outcomes in the intention-to-treat population.
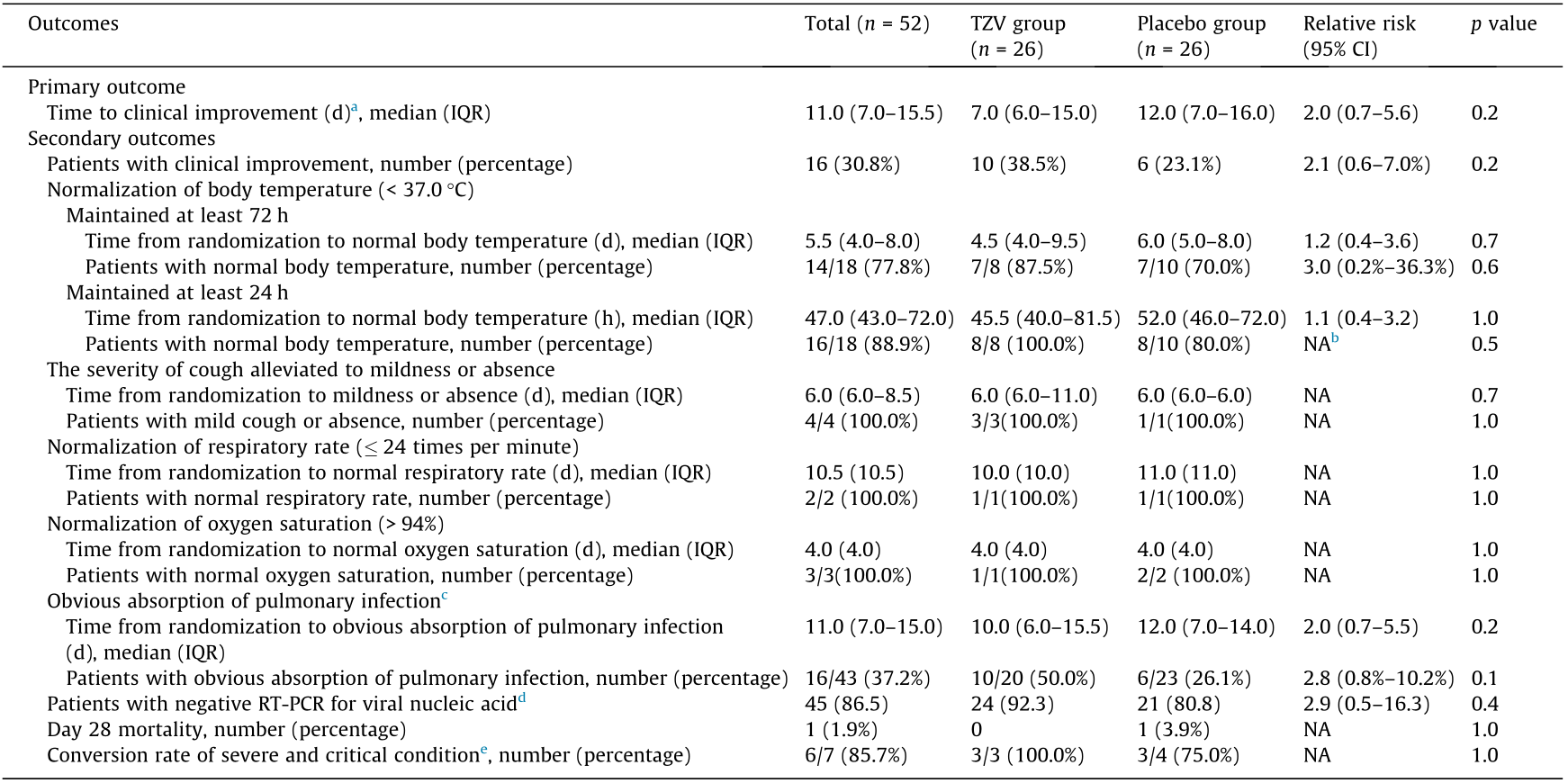
a Time to clinical improvement is defined as the number of days from randomization until normalization of the five points of body temperature, respiratory rate, oxygen saturation at the fingertip, alleviation of cough, and obvious absorption of pulmonary infection by chest CT, whichever came last. Normalization of body temperature is defined as a temperature less than 37.0 °C. A respiratory rate less than 24 times per minute is considered normal, and the oxygen saturation should be greater than 94%. Alleviation of cough is defined as the severity of cough changing to mildness or absent on a physician-reported scale of severe, moderate, mild, and absent. Obvious absorption of pulmonary infection is defined as an absorption area of more than 2/3 of the lesions by DICOM images on chest CT. All five points should be maintained at normal levels for at least 72 h.
b NA means not applicable.
c Obvious absorption of pulmonary infection is defined as an absorption area of more than 2/3 of the lesions by DICOM images on chest CT. DICOM image data from each site were uploaded to the imaging center of the First Affiliated Hospital of Harbin Medical University, and the final analysis was made by a pre-selected expert panel according to the unified standards.
d The throat swab viral nucleic acid test was repeated after 48 h if its result converted to negative.
e The conversion rate in severe and critical patients was assessed on a six-point scale. The patient being discharged from the hospital or the score decreasing by two points from baseline was considered to be conversion.
《3.2. Secondary outcomes》
3.2. Secondary outcomes
Clinical improvement was observed in ten patients of the TZV group and six patients of the placebo group in the intention-totreat population (38.5% versus 23.1%; RR, 2.1; 95% CI, 0.6–7.0; p = 0.2) (Fig. 2(a), Table 2). In patients whose body temperature was maintained at < 37 °C for 24 or 72 h, the rate of defervescence in the TZV group was higher than that in the placebo group (100% versus 80%, p = 0.5; 87.5% versus 70.0%, p = 0.6), and the median time to defervescence was 45.5 h versus 52.0 h, with p = 1.0 (for 24 h) and 4.5 d versus 6.0 d, with p = 0.7 (for 72 h), respectively.
The median time to absorption of pulmonary infection was 10 d versus 12 d (RR, 2.0; 95% CI, 0.7–5.5; p = 0.2; Table 2, Fig. 2(b)). Ten patients of the TZV group and six patients in the placebo group demonstrated significant absorption of pulmonary infection (50.0% versus 26.1%, RR, 2.8; 95% CI, 0.8–10.2; p = 0.1). The negativity conversion rate from a positive COVID-19 nucleic acid test to a negative result was 92.3% versus 80.8%, with p = 0.4 (Table 2, Fig. 2(c)). One patient who had been randomly assigned to the placebo group died over the course of the study (Table 2). There was no significant difference in the conversion rate from severe and critical condition to mild or ordinary condition between the two groups (100.0% versus 75.0%, p = 1.0).
《Fig. 2》
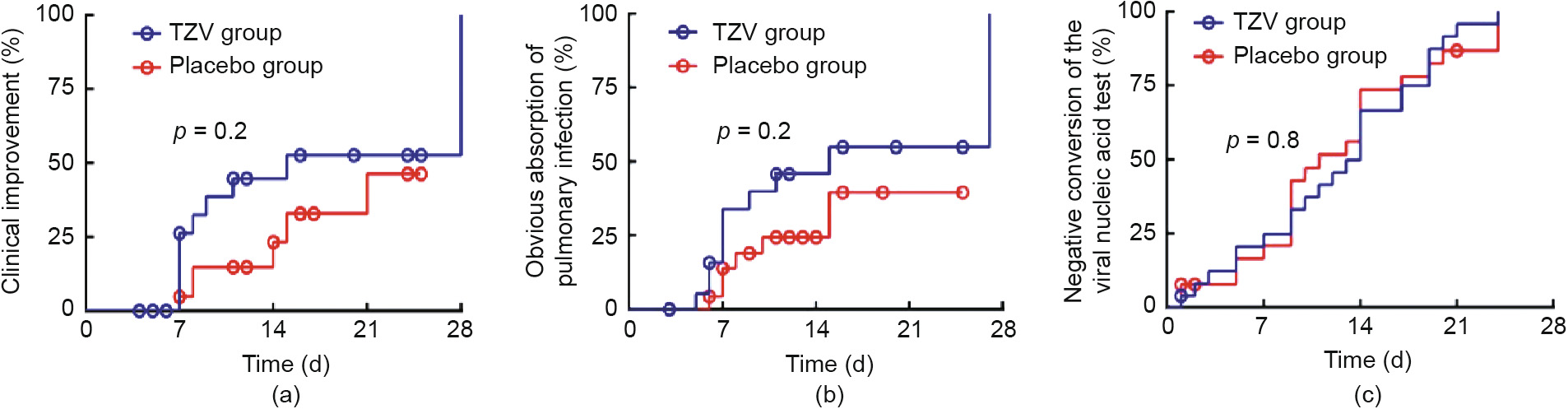
Fig. 2. The survival curves for clinical improvement, obvious absorption of pulmonary infection and the time of negative conversion of the viral nucleic acid test. (a) The survival curve with proportion of clinical improvement; (b) the obvious absorption of pulmonary infection by chest CT imaging; (c) the proportion with repeated negative viral nucleic acid test in 48 h by RT-PCR.
《3.3. Safety outcomes》
3.3. Safety outcomes
Adverse events were reported to be lower in six patients (23.1%) in the TZV group relative to ten (38.5%) in the placebo group (Table 3). Nausea and vomiting (11.5%), hypoalbuminemia (3.8%), granulocytopenia (3.8%), electrolyte disturbance (3.8%), and sour regurgitation and abdominal discomfort (3.8%) were reported in the TZV group, while anemia (11.5%), diarrhea (7.7%), and hypokalemia (7.7%) appeared in the placebo group.
Serious adverse events occurred in a total of nine patients, with four events in the TZV group and five events in the placebo group. One patient in the TZV group developed arrhythmia after taking the study drug. One death occurred in the placebo group, while nobody in the TZV group died. The patient who died during the trial was a 70-year-old man; his condition worsened on the day of enrollment, with his blood oxygen saturation dropping down to 83%. Upon adjusting the flow rate of oxygen to 7 L·min–1 , his blood oxygen saturation was maintained at 90% and his oxygenation index was less than 300 mmHg. Chest CT examination revealed a serious chest inflammation with 75% pulmonary shadow. The patient was transferred to the ICU the day after the enrollment day, and his condition continued to worsen with the development of severe respiratory and circulatory failure. The patient died on the third day after randomization (Table 3).
《Table 3》
Table 3 Adverse events, serious adverse events, and concurrent treatment.


《4. Discussion》
4. Discussion
Our randomized trial found that the combination of TZV and standard therapy was not associated with a statistically significant overall improvement of the clinical outcomes for patients with COVID-19 relative to a placebo. Such a negative result might be ascribed at least partly to the small sample size enrolled in the study.
A strength of this trial is that it was an add-on interventional study consisting of the Chinese standard therapy plus the study drug or placebo. This design ensured protocol compliance and maximized the enrollment of patients in this trial. There are also limitations. First, all assessments of clinical symptoms and signs were conducted by local investigators, who were primarily engaged in the emergent clinical affairs and who did not have trial Standard Operation Procedure training ahead of time. Second, no central laboratory was available for rapid and timely testing or the monitoring of every patient for sample shipping biosafety. Third, the sample size of our RCT was small. Because the RCT was designed and carried out in Heilongjiang Province and COVID-19 is a time-limited disease in this region of China, there were not enough newly infected patients for our trial with the planned sample size.
In terms of the overall efficacy of TZV in our study, the percentage of patients with clinical improvement in the TZV group was nearly twice as high as that in the placebo group, and the median time to clinical improvement was five days shorter with TZV than with a placebo. The rates of stable defervescence maintained for 24 and 72 h were both higher in the TZV group than in the placebo group. In fact, by the end of the follow-up period, the body temperature, respiratory rate, oxygen saturation, and cough recovered to normal in all participants, with the exception of the slow absorption of pulmonary infection as shown by chest CT. Furthermore, the beneficial effects of TZV were associated with a higher recovery rate of abnormal neutrophil percentage, lymphocyte count, and lymphocyte percentage in routine blood tests, and a higher serum level of CRP (Table S2). These results were consistent with reduced concurrent usage of antibacterial drugs, hydroxychloroquine, antifungal drugs, glucocorticoids, and non-steroid anti-inflammatory drugs for fever control (Tables 3 and S3).
The essential manifestation of clinical symptoms in COVID-19 patients comprises infection by SARS-CoV-2 and the development of pneumonia. As for the antiviral effect, the conversion rate of the negative virus test reached 92.3% in the TZV group, which was 10% higher than that in the placebo group. A higher absorption rate and a shorter absorption time in pulmonary CT in the TZV group indicated a reduction in the lower inflammatory lesions in the lung after treatment with antiviral agents. Moreover, less frequent usages of ordinary oxygen support and anti-tussive/anti-asthmatic/expector ant drugs to control symptoms occurred in the TZV group, demonstrating the beneficial effects of TZV on respiratory symptoms.
TZV treatment yielded higher recovery rates of abnormal serum levels of bilirubin, indirect bilirubin, total protein, albumin, and uric acid, as well as less usage of electrolyte solution and diuretics, indicating less damage in hepatic and renal functions. A high level of plasma fibrinogen is an important risk factor in the hypercoagulable state. Elevated D-dimer in the early stage of COVID-19 is closely related to the inflammatory response, while a sharp rise in D-dimer accompanied by respiratory failure suggests the possible generation of a "cytokine storm.” In this study, the recovery rate of D-dimer after TZV treatment reached 20.0%, while the recovery rate was 0 in the placebo group. Furthermore, the beneficial effect of TZV was manifested by less usage of antiplatelet and anticoagulant drugs.
In this study, the recovery rate of lactate dehydrogenase (LDH) was nearly twice as high in the TZV group as in the placebo group. A previously case study showed that cardiac damage is an important complication associated with COVID-19, even in patients without symptoms or signs of pneumonia [13]. Furthermore, TZV treatment was associated with less frequent usage of blood volume expanders, antihypertensive drugs, and drugs for dilating blood vessels to improve microcirculation in the TZV group. Thus, TZV may benefit COVID-19 patients by reducing cardiac damage. In summary, TZV may be beneficial in pulmonary, cardiac, renal, hepatic, and coagulable functions in patients with COVID-19 due to its antiviral properties. Larger studies are needed to confirm or refute these observations.
《5. Conclusions》
5. Conclusions
In this pilot trial, TZV showed potential for treating COVID-19 due to its antiviral effects by generally reducing inflammatory reactions and thus reducing damage to vital organs and reducing the need for therapeutic support.
《Acknowledgements》
Acknowledgements
We are deeply grateful to the front-line clinicians who participated in the study while directly fighting the epidemic. This study was supported by the Chinese Academy of Engineering Projects for COVID-19 (2020-KYGG-01-04) and Heilongjiang Province Urgent Project-6 for COVID-19. Data and safety monitoring board members of this trial included Kang Li, Yong Zhang, Songjiang Liu, and Yaohui Shi.
《Compliance with ethics guidelines》
Compliance with ethics guidelines
Xiaoke Wu, Kaijiang Yu, Yongchen Wang, Wanhai Xu, Hongli Ma, Yan Hou, Yue Li, Benzhi Cai, Liying Zhu, Min Zhang, Xiaoli Hu, Jingshu Gao, Yu Wang, Huichao Qin, Wenjie Wang, Mingyan Zhao, Xia Wu, Yong Zhang, Lu Li, Kang Li, Zhimin Du, Ben Willem J. Mol, and Baofeng Yang declare that they have no conflict of interest or financial conflicts to disclose.
《Appendix A. Supplementary data》
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.eng.2020.08.011.











 京公网安备 11010502051620号
京公网安备 11010502051620号




