
《1. Introduction》
1. Introduction
Since the initial reports on their generation in 2006 [1,2], induced pluripotent stem cells (iPSCs) have been investigated as promising cell sources for regenerative medicine [3–6], drug screening [6,7], and the food industry [8]. They serve as a renewable source for various cell types in the body due to their extensive proliferation and differentiation capacities, which are similar to those of embryonic stem cells (ESCs). In addition, as they are derived without embryo sacrifice, their generation is devoid of ethical concerns and their autologous transplantation does not cause immune-related complications. With these high expectations, iPSC investigations have advanced rapidly during the decade following their invention.
However, clinically or industrially relevant iPSC applications demand a considerable number of cells, and the methodology for stable mass production is in the developmental stages. For example, pancreatic islet transplantation requires at least 6 × 108 β cells per patient, and their generation requires approximately 0.6 m2 of culture surface [9], which is equal to 80 flasks that are 75 cm2 in size. In extreme cases, the preparation of 30% liver tissue requires 6 × 1010 hepatocytes per patient; these can be obtained from 60 m2 of culture area, which is equal to 8000 of such dishes. In addition, considering the differentiation process, more than twice the number of cells is required. According to research on cell therapy cost estimation, the assumed lot scale is approximately 1 × 1010–1 × 1011 cells per lot, which is equal to 100–10 000 vials per lot [10–12]. These requirements cannot be fulfilled via the laboratory-followed conventional adherence cultures involving manual operation. Therefore, it is necessary to develop a stable iPSC manufacturing process.
Various culture systems have been developed to produce large numbers of cells. Cell-based mass production systems have traditionally been developed for cell-derived products such as dyes, vaccines, and antibodies [13]. A different approach is essential for cell therapy applications because the cells themselves are the products. Although iPSCs are anchorage-dependent cells, they can also be cultured in suspensions via aggregate formation. Thus, iPSCs can be cultured using two different culture systems: adhesion-based culture system and suspension-based culture system. In addition, the scaffold-based approach is being developed to control the stem cell niche and stabilize iPSC quality.
During mass production, the downstream processes for cell filling and freezing are potentially critical for developing a stable iPSC production system. As the quality of cells changes in a timedependent manner during the production process, immediate filling and freezing is essential for stable production. A highthroughput filling and freezing system for cell therapy is still being developed; therefore, the lack of a suitable downstream process is currently limiting the lot scale. Although downstream processes have not been reported as widely as upstream processes such as expansion, the importance of downstream processes as the limiting factors for scaling up cell production is now being recognized.
In this review, we summarize the investigations on and developments in stable iPSC mass production systems in terms of upstream and downstream processes. First, we discuss the general limiting factors in iPSC culture, such as nutrient supply, waste removal, and the presence of growth factors. With respect to the upstream process, we introduce the features of and developments in culture systems based on adhesion culture, suspension culture, and scaffolding. We then discuss the downstream processes as a limiting factor for scaling up. Finally, we summarize recent progress on stable iPSC production and highlight the problems involved in the development of stable production systems for new industrial applications such as regenerative medicine.
《2. General limiting factors in iPSC culture》
2. General limiting factors in iPSC culture
《2.1. Nutrient supply and waste removal》
2.1. Nutrient supply and waste removal
As iPSCs mostly rely on glycolysis to meet their energy demands [14–17], a glucose feeding strategy is important for iPSC culture. Some studies have reported that maintaining a high glucose concentration in the medium enhanced proliferation and the expression of pluripotency markers in iPSCs [18,19]. The mechanism underlying these positive effects remains unclear; however, some reports indicate that glucose-induced hyperosmolarity plays an important role [19,20]. In addition, some reports have mentioned that glutamine oxidation is also essential for the survival of human PSCs [21]. Glutamine metabolism contributes not only to the synthesis of nucleotides and glutathione, but also to adenosine triphosphate (ATP) production by oxidative phosphorylation. During glucose depletion, glutamine oxidation is activated for the tricarboxylic acid (TCA) cycle to produce ATP. Based on these investigations, iPSC production requires glucose and glutamine control, and a stable supply of both is important for stable production (Fig. 1 [21]).
《Fig. 1》
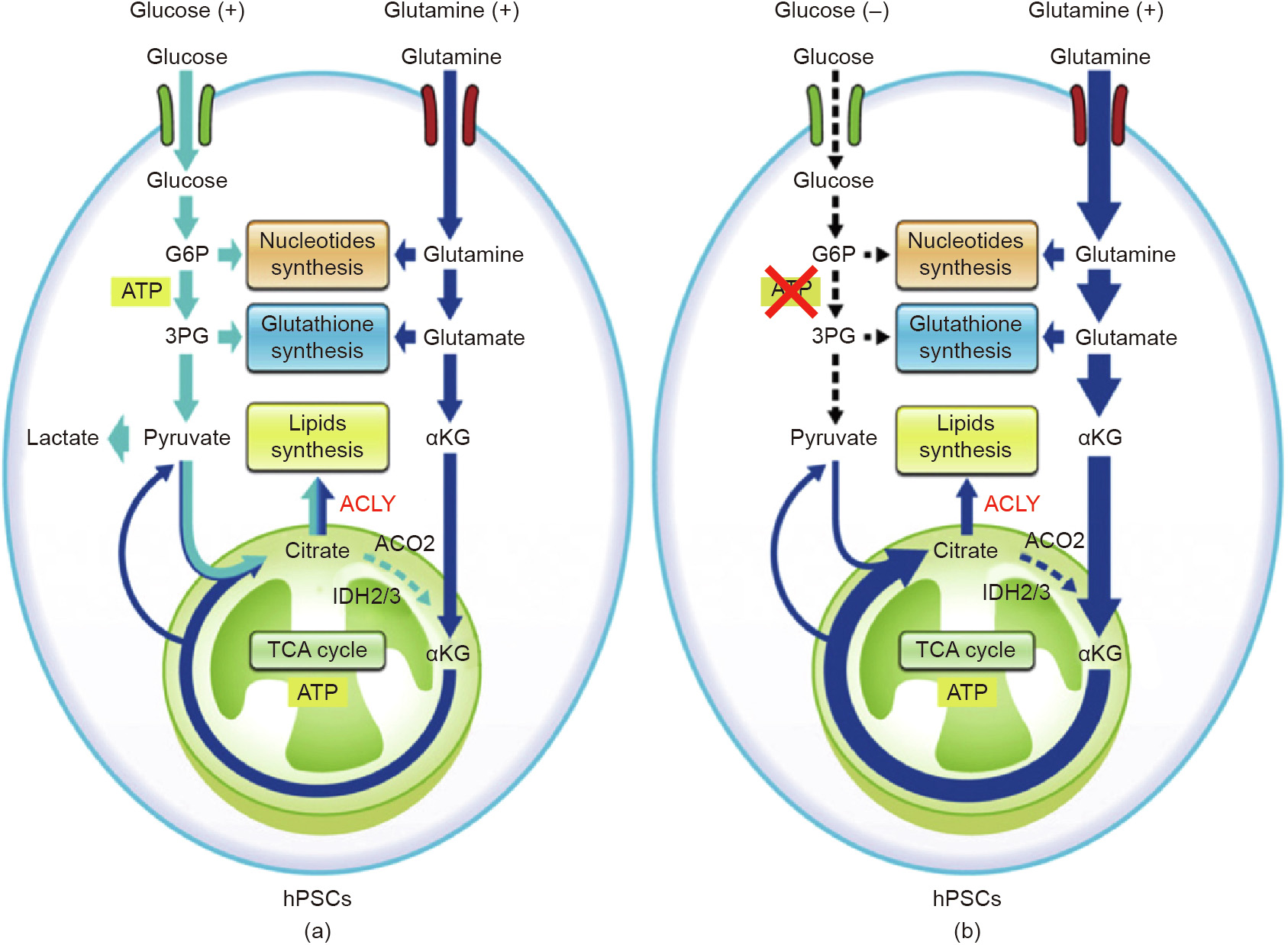
Fig. 1. Schematic image of glucose and glutamine metabolism in human pluripotent stem cells (hPSCs) in (a) glucose-existence and (b) glucose-depleted conditions. ACO2: aconitase 2; IDH2/3: isocitrate dehydrogenase 2/3; αKG: α-ketoglutarate; G6P: glucose-6-phospate; 3PG: 3-phosphate glyceraldehyde; ACLY: ATP citrate lyase. Reproduced from Ref. [21], with permission of Elsevier Inc., ©2016.
During culture, a considerable amount of lactate is secreted as a waste product of glycolysis. Lactate decreases the pH of the culture medium, and the resulting low pH causes iPSC death. In addition, previous research has shown that lactate decreases the growth rate, even in pH-controlled conditions [18].
These reports indicate that timely glucose supply and lactate removal are essential for a successful iPSC culture. Previous studies have also demonstrated that continuous feeding improved iPSC growth and pluripotency [22,23]. In particular, the use of a dialysis system for the effective and continuous transportation of nutrients and wastes is a successful technology for maintaining preferable culture conditions in terms of nutrients and waste concentration [23]. However, such systems need additional equipment, and thus require more space and complex operations. Therefore, it is essential to assess and meet the requirements of such systems during cell culture.
《2.2. Oxygen supply》
2.2. Oxygen supply
Oxygen is essential for cell culture, especially large-scale and high-density cultures. Some reports on two-dimensional cultures specify that cultivation on oxygen-permeable membranes resulted in the formation of a multilayer tissue and improved cell function [24–26]. However, in three-dimensional cultures, tissue thickness is limited by oxygen diffusion because the inner cells sometimes undergo necrosis due to a lack of oxygen [27].
As mentioned in Section 2.1, iPSCs produce their required energy via glycolysis, which does not require oxygen. Therefore, a low oxygen concentration (< 10%) is preferable for the maintenance of iPSCs, and has been reported to prevent iPSC differentiation [28,29]. However, oxygen depletion (< 1%) delays proliferation [29]. Thus, maintaining the oxygen concentration at 3%–10% is crucial for the effective production of iPSCs with a stable quality and in a stable quantity. Although oxygen is supplied from the top surface of the culture medium in most actual iPSC cultures, some investigations have utilized supplemental oxygen through a gaspermeable membrane, such as a closed system [30,31]. Sparging is also an effective method to supply oxygen; however, in the case of iPSC culture, sparging is currently rarely used because the medium flow caused by bubbles affects iPSC viability.
《2.3. Other supplements》
2.3. Other supplements
The addition of supplements such as growth factors is important in controlling cell fates such as growth, maintenance, and differentiation, and this holds true for iPSC culture. Rho-dependent protein kinase (ROCK) inhibitors such as Y-27632 and thiazovivin have important roles in the production of human iPSCs. In the past, human iPSCs could not survive if they were dissociated into single cells; therefore, when passaging human iPSCs, the cells should be harvested as small clumps. The passaging of human iPSCs is very difficult and requires expertise. A previous study demonstrated that the death of dissociated single human iPSCs is caused by ROCK-dependent apoptosis [32]. Recently, the addition of a ROCK inhibitor was shown to significantly improve the cell survival rate after passaging, and simplified the passaging process. ROCK inhibitors have other effects on iPSCs beyond improving the survival rate after passaging. A previous report mentioned the following additional effects of ROCK inhibitors on human iPSC cultures: improving the efficiency of cryopreservation, supporting undifferentiated growth, and enhancing differentiation [33]. Thus, ROCK inhibitors are useful in various situations for the stable production of human iPSCs.
Basic fibroblast growth factor (bFGF) is essential for pluripotency and the self-renewal of human iPSCs [34–36], so it is generally added to iPSC culture media. However, the lack of thermal stability in bFGF necessitates frequent medium changes, which increases the cost of mass production [37]. Nevertheless, several approaches, such as the sustained release of bFGF and improvement in bFGF thermal stability [37–39], have been suggested to decrease the frequency of the medium changes required for effective bFGF supplementation.
Transforming growth factor-β (TGF-β) superfamily proteins, such as TGF-β proteins, activin, nodal, and bone morphogenetic proteins (BMPs), also have an important role to play in maintaining the pluripotency of iPSCs. TGF-β1 has been shown to contribute to maintaining the pluripotency of human iPSCs [40,41]. Nodal and activin A activate the same receptor and signaling to suppress human iPSC differentiation and maintain pluripotency. However, a high concentration (approximately 100 ng·mL-1 ) of activin A also promotes the differentiation of human iPSCs into mesoendodermal cells [41,42]. Therefore, the activin A concentration should be maintained at below 50 ng·mL-1 in order to maintain the pluripotency of human iPSCs. BMP4, which belongs to the TGF-β superfamily, induces the differentiation of human iPSCs, even though BMP4 maintains the pluripotency of mouse iPSCs [43,44]. BMP4 can be secreted from cells and detected in serum replacement based culture medium. The antagonist noggin co-works with bFGF to suppress the differentiation induction of BMP4 and maintain pluripotency [35].
Finally, researchers have reported that various factors, including culture medium, substrate, and dissociation methods [45], affect cell characteristics not only in single-passage culture, but also in multiple-passage culture [46]. These fluctuations of quality that prevent stable production remain as problems to be solved, as mentioned in Section 5.
《3. Expansion process》
3. Expansion process
The currently developed iPSC mass production systems are based on one of the two main types of culture system: adhesionbased culture system and suspension-based culture system. An exception is the scaffold-based culture system, which involves both an adhesion-based culture system and a suspension-based culture system.
《3.1. Adhesion-based culture system》
3.1. Adhesion-based culture system
Adhesion-based culture is a conventional method for culturing mammalian cells in laboratories. In traditional adhesion-based culture, cells are seeded onto a substrate containing feeder cells such as mouse embryonic fibroblasts, which causes the crosscontamination of cells. Recently, various extracellular matrix (ECM) coatings have been developed for feeder-free iPSC culture. The ECM should contain an Arg–Gly–Asp (RGD) motif such as that in Matrigel, laminin, or vitronectin because the cell-substrate adhesion of iPSCs is mainly controlled by integrin [47]. Initially, animal-derived ECM such as Matrigel was used; however, to ensure biosafety, animal-free ECM comprising fragments of ECM molecules, such as the laminin-511 fragment [48,49] and vitronectin fragment [50], have been developed recently.
For mass production by means of adhesion-based culture, a large culture surface is required. Therefore, the use of methods to increase the culture surface in a vessel, such as stacking plates, is an important approach to increase productivity [51,52]. However, it is difficult to handle vessels with a large culture surface, so they are considered to be a secondary option when a microcarrier cannot be used.
Automating the culture operation is another approach to increase productivity and ensure stable quality [53–58]. Many companies around the world have developed mechanization culture systems that contain conduits for feeding medium and removing waste medium, as well as robotic arms to handle the experimental apparatus such as culture vessels, pipettes, and tubes. Stable operation and the possibility of parallel production are the main advantages of such mechanization systems. In fact, the possibility of parallel operation via a flexible culture platform has been reported [58]. However, despite advances in hardware development, software applications for the optimization of operations and monitoring cell quality are still being developed. Cell culture operations are complicated and vary with cell type; therefore, it is important to understand the effect of operations on the cell culture outcomes and to optimize culture operations to ensure stable production in adhesion cultures.
One advantage of adhesion-based culture is the easy observation of cell morphology. In the case of iPSC culture, including maintenance and differentiation, daily determination of cell state based on morphology is important for quality control. Hence, monitoring the morphology of iPSC colonies is an effective approach to check their quality and stability during mass production. These morphological determinations are conducted by experienced specialists. Moreover, the skill and knowledge of such specialists in morphological determination depend on operator experience, and it is difficult to train other personnel to attain a similar level of expertise. At present, image-based analyses for evaluating the quality of iPSCs are being developed [59–61]. Recently, deeplearning studies have been applied to the image-based quality control of iPSC products in adhesion-based culture [62,63]. These approaches attempt to evaluate the quality of iPSCs and iPSCderived cells without using conventional biological quality checks such as immunostaining, which requires skill and is unstable due to the fluctuating quality of reagents such as antibodies. Although thousands of teaching images are required to establish a reliable tool, these technologies should be powerful tools for monitoring the quality and stability of iPSC production.
In summary, adhesion-based culture is a conventional cell culture method, and the knowledge and experience from laboratory experiments can be utilized to ensure optimal production. Although scalability is limited in such culture systems, parallel production using mechanized production systems can overcome this hurdle. To realize stable mass production by means of an automated system, investigations are necessary on how cell characteristics are affected by each operation, including transportation, pipetting, centrifugation, and seeding. To understand the effect of such operations, biological investigations on mechanotransduction to mechanical forces [64,65] might be helpful. In addition, research on extraction of critical parameter from operation affecting cell quality in terms of chemical engineering is important when designing an automated culture operation.
《3.2. Suspension-based culture system》
3.2. Suspension-based culture system
Suspension-based cultures have been developed as scalable culture systems for bacterial [66], plant [67], and animal cells, and are used to obtain various biological products such as fermented foods, medicines, and cells themselves. Unlike adhesion-based cultures, suspension-based cultures do not require an adhesion surface, thereby enabling the use of simpler and easily scalable vessels. Several studies have reported the use of suspension-based culture for iPSC and ESC expansion [66–73] and differentiation [71–74].
As iPSCs require attachment, they form aggregates in suspension-based cultures. However, there is a size limitation for aggregate formation because of various factors, including the limited transfer of nutrients and oxygen into the aggregates [27,75] and ECM accumulation, depending on the cell lines. Previous research has shown that some cell lines form ECM with a shell-like structure, which packs the cells and prevents cellular growth [76,77]. It was also reported that the lack of ECM caused cell death and unstable aggregate growth [78]. Therefore, the characteristics of the iPSC line should be considered before designing a suspension-based culture system.
Because of these limitations, aggregation control is important during iPSC suspension-based culture. In particular, the formation of fewer aggregates in the early stage results in lower growth [76]. Therefore, an ideal scenario would encompass the generation of many size-controlled aggregates with cell–cell contact sufficient for stable growth. Some strategies have been suggested to control aggregation in the early stages of the suspension-based culture. One popular strategy is to use a microwell for the preparation of uniform aggregates [79]. Other possible suggestions involve modifications in the culture medium [80] or culture vessel [31] to limit cell aggregation. Furthermore, it has been reported that breaking up large aggregates into smaller ones by adding molecules that degrade cell–cell contact in the later stage of culture is an efficient approach to improve iPSC growth [30,81,82].
Since iPSCs do not require a high oxygen supply, bioreactors for iPSC expansion do not require strong agitation, unlike conventional bioreactors for microorganisms. However, this does not mean that bioreactors for iPSCs do not require any agitation. As mentioned above, iPSCs form aggregates in suspension-based culture, which can easily sediment and accumulate on the bottom of the bioreactor; therefore, agitation is required to prevent these problems. On the other hand, during suspension-based culture, cells are exposed to shear stress resulting from medium flow due to agitation. This shear stress affects iPSC viability [71] and differentiation [88]. Therefore, an optimal level of agitation that prevents sedimentation while being gentle enough to avoid causing cell damage is important for successful suspension-based culture in bioreactors. An estimation of the shear stress experienced by the cells is necessary when designing a bioreactor for iPSC suspension-based culture. As it is difficult to measure shear stress on cells directly, the use of computational fluid dynamics to estimate the shear stress has been suggested [89–91]. These computational sciencebased approaches are helpful for understanding the effect of shear stress on iPSCs and for developing optimal bioreactors for iPSC suspension-based cultures.
Various bioreactors have been developed for iPSC suspensionbased culture. The most popular suspension-based culture system for iPSCs is the spinner flask system, which has been widely researched [68,70,71,73,74]. In a conventional spinner flask system, agitation is operated by an impeller to realize a high level of oxygen transfer; therefore, the agitation rate is high (i.e., approximately greater than 100 revolutions per minute (rpm)) [66]. In contrast, agitation should be minimized when culturing iPSCs (i.e., kept at approximately below 60 rpm) to keep the cells floating with minimum shear force, because iPSCs do not require a high level of oxygen transfer. In spinner flasks, the culture medium is agitated directly, resulting in good mixing. However, the shear force from the impeller causes cell damage. A research group investigated the shaking culture system [31,72,83]. In a shaking culture, the culture medium is agitated indirectly by shaking the vessels. Due to the absence of an impeller, the shear stress on cells is lower in a shaking system than in a spinner flask. In addition, the culture vessel for a shaking system can be simplified, making it possible to scale up for mass production and easily create a closed system to maintain aseptic conditions. In a shaking culture, iPSC aggregates tend to sediment to the bottom of the vessel due to the weak agitation; thus, further design is required to prevent such sedimentation. As a system that is similar to shaking vessels, rotating-wall vessels can realize culturing with microgravity [84]. Previous studies have demonstrated that the microgravity produced by rotating-wall vessels enhanced viability, proliferation, and differentiation efficiency [85,86]. As an extreme case of a culture system with microgravity, a bi-directional rotating vessel system was developed, which realized pseudo zero gravity [87]. As mentioned above, there are various types of bioreactors, and it is important to choose according to the purpose (i.e., expansion or differentiation) and scale of the culture.
《3.3. Scaffolds-based culture system》
3.3. Scaffolds-based culture system
The third possible culture system involves culturing on scaffolds such as microcarriers and microcapsules. Microcarriers provide larger adherent surfaces than those provided by conventional adhesion-based cultures, and are widely used for the mass production of adhesion-dependent cells such as mesenchymal stem cells. As iPSCs are also adhesion dependent, the use of microcarrier-based culture systems for the mass production and differentiation of iPSCs has been reported [88,92–94]. In general, unlike the cells in static adhesion-based cultures, the cells on microcarriers are suspended in a stirring vessel and exposed to shear stress, which affects cell viability and differentiation (Section 3.2). In addition, when harvesting cells after expansion, a separation microcarrier from the cell suspension is required. This process potentially limits the scale of the culture.
The selection of a microcarrier is important for successful iPSC culture on a microcarrier. According to previous investigation, size, coating, and surface charge are important properties to consider for iPSC expansion [92]. According to Ref. [92], use of a small microcarrier (smaller than 100 μm) results in a low cell yield. In addition, the iPSCs barely attached or grew on a microcarrier with a negatively charged surface; thus, a positively charged surface such as an amine-conjugation is required. Coating the ECM improves cell growth on microcarriers. In Ref. [92], Matrigel and laminin coating were found to be effective to improve cell growth. To solve the problem of separation from the microcarrier, novel microcarriers that are dissolvable by enzymatic treatment were recently developed [94]. By digesting the microcarrier after expansion, it is possible to skip the separation process.
Microencapsulation, another approach for the scaffold-based culture of iPSCs, enables the protection of cells from shear stress, aggregation control, and the preparation of preferable structures via bioprinting [95]. Some reports indicate that encapsulation can prevent differentiation and preserve the pluripotency of iPSCs [96,97]. Moreover, the microcapsule can be modified to control the stem cell niche [98]. Although these advantages indicate the possibility of stable mass production of iPSCs, the collection of iPSCs from capsules after culture is a barrier to the application of such scaffold-based production processes.
Various encapsulating materials have recently been investigated, such as polyethylene glycol (PEG), agarose [99], and hyaluronic acid [100]. The most popular encapsulating material is alginate hydrogel. Alginate immediately forms a hydrogel when it is dropped into a divalent cation solution such as a solution of calcium, barium, and iron ions. Thus, the encapsulation of cells can be easily realized by dropping a cell suspension with sodium alginate solution into a divalent cation solution. Furthermore, it is easy to digest alginate hydrogel by soaking the capsules in a chelate (e.g., ethylenediaminetetraacetic acid (EDTA) and citric acid) or alginate lyase solution. As a further treatment for capsules, because alginate hydrogel capsules have a negative charge on their surface, the alginate hydrogel can be covered with a poly-ion complex membrane by soaking the hydrogel capsule in a poly-cation such as poly-L-lysine and chitosan. After forming a poly-ion complex, hollow capsules with a liquid core can be prepared by soaking the coated capsule in a chelate solution [96,97]. Previous investigations have reported that the simple encapsulation of pluripotent stem cells (PSCs) into alginate resulted in cell leakage from the capsule and growth outside of the capsules; therefore, forming a polyion complex membrane is essential for the expansion of PSCs in capsules [96,97,101].
《4. Filling and freezing processes》
4. Filling and freezing processes
In addition to the upstream parts of the iPSC production process, the downstream processes that follow the expansion processes must be considered during iPSC culture investigations. These downstream processes, which include the filling and freezing processes, are essential for packaging the expanded cells.
《4.1. Before freezing (filling)》
4.1. Before freezing (filling)
During the filling process, dispensing 0.5–2.0 mL of the cryoprotectant-containing cell suspension into vials is a timeconsuming task that affects the quality and viability of iPSCs after cryopreservation due to the toxicity of the cryopreservation medium. Due to this issue, scaling up is limited to a level at which the complete filling process, including the transfer from the culture system to the filling system, can be completed within 1 h. Given the time limitation associated with the conventional manual filling procedure, the estimated scale of expansion is approximately 1–5 L, which can produce 1 × 109 –1 × 1010 cells per batch. This means that thousands to ten thousands of vials need to be filled with cell suspension from the same batch. During the filling, cells decay in a time-dependent manner, causing an unignorable difference in quality within the same batch. A previous report indicated that 1 h is the optimal time to ensure the maximum viability of iPSCs in cryopreservation medium [102]. Thus, to obtain the required lot scale, the development of high-throughput filling processes and parallel production systems is essential. This limitation is mainly due to the dimethyl sulfoxide (DMSO) in the cryopreservation medium. It has been reported that DMSO causes protein aggregation [103] and damages the mitochondrial integrity and membrane potential [104]. In the case of cryopreservation of iPSCs and ESCs, it has been reported that exposure to DMSO decreased the pluripotency genes [105,106] and promoted differentiation markers [106]. Therefore, cryopreservation methodologies with a DMSO-free cryopreservation medium have been investigated. Previous studies have shown that ethylene glycol is a promising alternative to DMSO, with lower toxicity and high efficiency in vitrification [107,108]. In addition, the use of a freezing medium consisting of ethylene glycol, sucrose, and carboxylated poly-L-lysine has been shown to improve cell viability after slow vitrification by inhibiting ice crystallization [109].
《4.2. Freezing (cooling down)》
4.2. Freezing (cooling down)
Freezing is one of the critical operations involved in cell production. Conventionally, iPSCs are cryopreserved by means of vitrification—that is, immediate freezing with liquid nitrogen in highly thermoconductive containers such as open-pulled straws. This technique was developed for the preservation of bovine ova and embryos and is utilized for PSC preservation. Vitrification prevents the formation of crystals that cause cell damage during freezing; thus, it is a successful method and realizes a high recovery rate (more than 75%, compared with 5%–10% after slow-cooling) on a laboratory scale. However, the cryopreservation medium used for vitrification generally has a high concentration of cryoprotectants such as DMSO and ethylene glycol, giving the medium high osmolality and toxicity. Therefore, rapid thawing is important for the successful recovery of vitrified cell samples. This is accomplished by immersing the vitrified cell samples into a pre-warmed culture medium. Although vitrification is a successful method that is widely used in laboratories, it has certain limitations in terms of the stable mass production of PSCs, such as difficulty controlling the temperature and contamination due to direct contact with liquid nitrogen. A previous report suggested the novel design of a culture plate with detachable wells and screw caps for the storage of adherent human ESCs by vitrification, which may assist in the development of automated systems for handling bulk quantities of cells [110].
Although slow freezing has shown poor performance in the past [111], the development of cryopreservation medium [112,113] and advances in methodologies for freezing with a slow-cooling system [114] have improved the performance of these systems and the associated cell productivity. If the speed used for slow cooling is too fast, rapid freezing will cause intracellular ice crystal formation. These intracellular ice crystals disrupt cellular organelles and membranes and lead to cell damage and death during thawing and re-seeding [115,116]. On the other hand, freezing that is too slow results in the removal of intracellular water due to osmotic pressure [115,116], which causes cellular dehydration and shrinkage, resulting in the disruption of organelles, the plasma membrane, and the cytoskeleton, and finally causing cell death. Thus, in a slow-cooling system, the cooling rate should be slow enough to prevent intercellular ice formation yet fast enough to prevent intracellular water removal. The reported optimal cooling rate is 0.5–1.0 K·min-1 [114,116,117]. In the slow-cooling method, the temperature is controlled and gradually decreased to a final temperature (lower than –80 °C) [118]. A programmed deep freezer is used for slow cooling, and various temperature control procedures have been reported. However, when considering scaling up, a simple cooling program is important to permit temperature controllability during bulk cooling. This method is feasible for scaling up, but the acceptable difference in temperature (i.e., the temperature robustness of the process) remains unclear despite several reports on cryopreservation. This lack of temperature robustness will affect the scalability of the freezing process because scaling up causes temperature variation due to heat conduction.
At present, a deep-supercooling method is being developed as another approach using rapid cooling [119,120]. In this method, cells are preserved without freezing at a higher temperature compared with conventional cryopreservation. The difficulty with this method is to maintain supercooled water in an unfrozen state, as it readily forms ice crystals from various stimuli. A recent study showed that sealing the surface with immiscible liquid improved stability, and enabled the researchers to store human red blood cells for 100 d at –7 to –10 °C [120]. After further development, this method will be a promising candidate for storing cells as a final product.
To summarize, the downstream process remains a potential bottleneck for scaling up iPSC production, and it is essential to develop novel approaches for optimal downstream processing. To do so, it is necessary to investigate the effects of various conditions—such as suspension time in cryopreservation medium and temperature difference—on cell viability and quality.
《4.3. After freezing (storage, transportation, and thawing)》
4.3. After freezing (storage, transportation, and thawing)
After freezing, the frozen cell suspension is generally stored in a cryopreservation tank with liquid nitrogen (at approximately –196 °C in the liquid phase and –170 °C in the gas phase), as in conventional cell lines. In particular, a temperature below –130 °C is ideal for long-term cell storage. This is because liquid free water does not exist at temperatures below –130 °C, and the crystalline or glassy state has a high enough viscosity to prevent the effects of diffusion and stop biological time completely. Although many cell lines are stored at temperatures ranging from –70° to –90 °C for months or years, biological time is not stopped in such instances, but rather is slowed. Thus, biological reactions affecting cell viability and characteristics still occur and these effects will accumulate. Indeed, a previous study showed that the viability of liver cells decreased during storage at –80 °C but was maintained for a year at –170 °C [121]. During cell production, the temperature fluctuates as a result of various operations, such as during transportation from the freezer to storage. In addition, the transportation methodology for frozen cell samples is still developing. Previous research reported that a repeating temperature fluctuation during storage decreased cell viability after thawing [122]. The effect on frozen cells of temperature fluctuation and acceleration of container during transportation is unclear. Thus, understanding the robustness of the quality of frozen cells to these fluctuations is necessary in order to develop a stable cryopreservation system.
In addition, an effective methodology to transport frozen cells has not yet been developed. Aside from suitable thermal management, the effects of transport vibration on frozen cells are still not well understood and must be considered. With an understanding of the robustness of frozen cells to temperature change and force, the stable transportation of cell products can be realized.
Thawing cells is another critical operation that affects cell quality. Traditionally, it is important for successful recovery to thaw cells quickly at 37 °C and to avoid overheating [116]. Slow thawing has been reported to cause cell damage due to ice crystal reformation. Although some work has been accomplished in this field, further understanding of the decay that occurs when heating cells and the optimization of this process is necessary in order to obtain stable and successful results with cell products.
《5. Future perspective: Unresolved barriers to stable production》
5. Future perspective: Unresolved barriers to stable production
As mentioned above, it is necessary to develop a scalable process from the expansion process to the filling and freezing processes. Despite the investigations that have been done on PSC at the laboratory scale, the stable mass production of iPSCs is still developing. One of the major barriers to mass production is that the operations in iPSC production are complicated and are performed manually, potentially resulting in fluctuations in cell quality due to fluctuations in manual handling. In addition, as mentioned in Section 2.3, cell characteristics are mutated in a time-dependent manner during culture [46,123]. Recent analyses have demonstrated that the characteristics of cells, including gene expression and drug response, have wide heterogeneity across laboratories, even within the same cell strain [124,125]. Unlike the production of cellular products such as antibodies, this heterogeneity cannot be ignored and should be minimized because the cells themselves are the products in the iPSC production process. In order to avoid such heterogeneity, it is essential to establish an appropriate quality-control method, which will include the development of evaluation methods and setting up appropriate evaluation parameters.
It is also necessary to develop a reproducible production process. As mentioned in Section 3.1, automation is promising approach to realize high reproducibility of culture operation for stable production. Although various industries have developed automated culture systems, the conversion of operations from manual handling to automated handling is still developing. This development requires an understanding of what the critical operations and parameters are in cell culture. For developing processes such as automated cell culture systems, chemical engineering knowledge is helpful. In terms of chemical engineering, various parameters have been utilized to optimize the cell production process. For example, the Reynolds number—that is, the ratio of inertial forces to viscous forces—has been developed to predict flow patterns even under different flow conditions. When developing a stable production process for iPSCs, such parameters are helpful for predicting the result of an operation before actually performing it. In a recent study, a group specializing in transport phenomena demonstrated that the Froude number, which is the ratio of the flow inertial centrifugal force and the gravitational body force, can be used to predict the heterogeneity of cells after shaking during the seeding process [126]. These parameter designs and performance indices are necessary for developing a stable production method for iPSCs.
In terms of quality control, both biological analyses and statistical strategies—including deep learning—are expected to develop, as mentioned in Section 3.1. Deep learning is rapidly progressing in various fields including bioinformatics, computational biology, and medical applications [127,128]. These analyses can realize quality checking without destruction and permit real-time monitoring via time-lapse capture by microscope. However, such analyses also tend to be ‘‘black box” models, so support with biological experiments is important in order to establish an effective deep-learning system.
The other difficulty in developing stable mass production of iPSCs is that the effects of operations on cells are indirect. For example, during the detachment of cells by pipetting, the pipetting operation can be defined by flow rate; however, the effect of this operation on cells occurs in the form of shear stress. In other words, the parameter that can be adjusted during operation and the parameter that actually affects the cells are different. Thus, further investigation on how to translate the operation parameters into the parameters that actually affect the cells is important in order to develop a stable mass production process. Such investigations require not only biology, but also various engineering fields such as mechanics, fluid dynamics, and computational engineering.
《6. Conclusions》
6. Conclusions
In this review, we discussed the development of and hurdles in the upstream processes such as expansion and the downstream processes such as filling and freezing involved in iPSC production. We presented the strengths and weaknesses of the adhesion-, suspension-, and scaffold-based culture systems and discussed the downstream processes as a possible bottleneck for iPSC mass production. Although iPSCs have been researched for several decades, the industrialization of iPSC production has not yet been achieved. To ensure stable iPSC production, it is essential to investigate the manufacturing process to determine the evaluation factors for cell quality, identify the inputs that affect cell quality during operations, and understand the mechanisms underlying these effects. For these investigations, it is necessary to design experiments to study iPSC production from various points of view such as biology, mechanical engineering, physics, and computational engineering. The problems mentioned above do not only apply to the production of iPSCs, but are also issues in the production of other common cell products such as mesenchymal stem cells. Thus, engineering approaches to solve these problems will be useful for the stable production of various cell products including regenerative tissue and artificial meat.
《Acknowledgements》
Acknowledgements
This research was supported by Japan Aency for Medical Research and Development (AMED; JP20be0604001).
《Compliance with ethics guidelines》
Compliance with ethics guidelines
Ikki Horiguchi and Masahiro Kino-oka declare that they have no conflict of interest or financial conflicts to disclose.











 京公网安备 11010502051620号
京公网安备 11010502051620号




