
《1. Introduction》
1. Introduction
Acute myeloid leukemia (AML) is the most common form of leukemia in adults, accounting for approximately one-third of all leukemias worldwide. In the United States alone, over 20 000 patients are diagnosed annually with AML, and it is estimated that the overall worldwide incidence of AML is 350 000 [1]. The median age for adults has been steadily increasing over the past three decades and is now approaching 70 years, partly due to the greater readiness of physicians to diagnose AML in older adults. Of all leukemias, AML has the lowest survival rate, with only 28% of adults surviving longer than five years [2]. The overall incidence of AML in the United States is 3.4 patients per 100 000 population, with 1.2 patients per 100 000 at age 30 years but more than 20 patients per 100 000 population at 80 years [3].
Therapy for AML has been consistent; for almost five decades, the backbone of therapy has depended on the use of an anthracycline and cytarabine—the so-called 7 + 3 regimen [4]. Although the 7 + 3 regimen was initially developed in 1973, its use only became widespread a few years later, with the wide availability of platelet transfusions that emerged after the landmark discovery of the ability to store platelets in 1975 [5]. The main use of the 7 + 3 regimen has been in induction therapy, aiming for the achievement of a complete remission (CR). For decades, the attainment of CR has been considered a sine qua non for the achievement of long-term survival [6]. Crucial studies in the early 1980s established that long-term survival required the administration of post-remission therapy, which consisted of high doses of chemotherapy, autologous hematopoietic stem cell transplantation (ASCT), allogeneic hematopoietic stem cell transplantation (allo-SCT), or protracted maintenance therapy [7]. Over five decades, the long-term survival of AML has increased considerably, particularly in younger individuals [8] (Fig. 1).
《Fig. 1》

Fig. 1. Long-term survival of patients < 60 years old with newly diagnosed AML. Data from successive studies of the Eastern Cooperative Oncology Group (ECOG) and updated from Ref. [8]. N: number of patients; OS: overall survival.
The first successful targeted therapy for AML occurred with the recognition of all-trans retinoic acid (ATRA), which fuses directly with the retinoic acid receptor (RAR) alpha and is now the standard of care for acute promyelocytic leukemia (APL). This therapy transformed a previously highly aggressive form of AML into a subtype of AML that could be cured in more than 70% of patients [9]. This advance was followed by the discovery of imatinib mesylate for the treatment of Philadelphia chromosome-positive chronic myeloid leukemia (CML), in which this agent was employed as a unique target for the BCR–ABL oncogene in transforming a highly malignant disease into one that allows for long-term survival in almost all patients [10].
Unfortunately, other targeted agents for myeloid leukemias have not had the same dramatic success as ATRA and imatinib. Both of these agents were directed at a disease powered by a single driving mutation, t(15;17) in APL and t(9;22) in CML. Most AML subtypes have multiple driving mutations, making specific targeted therapy more elusive and less likely to be effective as monotherapy.
One of the most important targeted agents for AML has been venetoclax, which attaches to the Bcl-2 protein. Initially approved with much enthusiasm for the treatment of chronic lymphocytic leukemia, venetoclax was soon revealed to be one of the most potent agents for use in AML; it has now been incorporated into the standard of care, particularly for older patients with newly diagnosed AML, provided that it is given with either azacytidine [11] or low-dose cytarabine [12]. Also of interest are the recently approved inhibitors of isocitrate dehydrogenase (IDH). Mutations in IDH have been found in 15%–18% of patients with AML. Inhibitors for both IDH1 (ivosidenib) and IDH2 (enasidenib) have proven to be highly effective agents for the treatment of advanced AML [13,14]. The Hedgehog pathway inhibitor, glasdegib, was also recently approved, in combination with cytarabine, for older adults with newly diagnosed AML [15].
One of the most important classes of inhibitors arose out of the discovery of several kinase inhibitors acting specifically, or more broadly, against mutant forms of the FMS-like tyrosine kinase 3(FLT3) activating mutations. Since these mutations—which are among the earliest molecular abnormalities described in AML— are common, a major effort has been underway to target these groups of patients. Several agents with varying specificity are in clinical use and are currently being investigated for therapeutic application. The frequency of FLT3 mutations in AML has stimulated the investigation into a number of tyrosine kinase inhibitors (TKIs) in an effort to disrupt the oncogenic signaling driven by FLT3. Midostaurin, a targeted first-generation FLT3 inhibitor, is the first of nine new agents that were very recently approved for AML, after a hiatus of four decades [16]. Therefore, we selected this group of FLT3 inhibitors for discussion in this article as the paradigm of targeted therapy for AML, representing multiple stages, from single small trials to widespread multi-institutional international trials and clinical use. All stages of therapy will be discussed in detail, including induction, consolidation, and maintenance— both post chemotherapy and post-allogeneic transplantation. The impact of these inhibitors on the prognosis of AML will be considered, and the multiple agents that are used will be discussed in considerable detail in this review, with an emphasis on their clinical application.
《2. FLT3 inhibitors》
2. FLT3 inhibitors
As antineoplastic therapy becomes increasingly selective, any recognized mutation is rigorously studied as a potential target for therapy. An activating mutation in the transmembrane tyrosine kinase FLT3 was first identified in AML patients in the mid-1990s [17]. Subsequent research showed that approximately one-third of AML patients harbored the mutation, making it one of the most prevalent genetic alterations in the disease [18–21].
The FLT3 is a tyrosine kinase receptor, as are the KIT and platelet-derived growth factor receptors (PDGFRs) [22]. It is located on chromosome 13q12 and has an extracellular region, a juxtamembrane domain (JM), and a tyrosine kinase domain (TKD). The receptor is expressed on normal hematopoietic stem cells and plays an important role in the proliferation, differentiation, and survival of stem cells. As in other tyrosine kinase receptors, mutations in the FLT3 receptor result in constitutive activation of the kinase via autophosphorylation and the activation of several signaling downstream pathways. This causes rapid cell proliferation and reduced apoptosis [23,24]. Two distinct FLT3 mutations have been identified in AML patients: The first is an internal tandem duplication (ITD) found in the JM, which is referred to as the FLT3-ITD mutation, and the second is a missense point mutation within the TKD, which has been coined as the FLT3- TKD mutation [25] (Fig. 2). The more prevalent mutation is the FLT3-ITD mutation, which is present in approximately 25% of all newly diagnosed AML patients; in comparison, it is estimated that 7%–10% of AML patients harbor the FLT-TKD mutation. Both mutations result in constitutive activity of the FLT3, but only the FLT3- ITD has been shown to be a driver mutation with clear prognostic implications [26]. FLT3-ITD was soon shown to be associated with rapid relapses and shorter overall survival (OS) in AML patients, particularly in those with a higher allelic burden, which has been arbitrarily defined as a mutant-to-wild-type ratio greater than 0.51–0.78, depending on the study [18,27]. The implications of the FLT3-TKD mutation have yet to be completely elucidated, and while some studies showed weak associations with clinical outcomes, others did not [28].
《Fig. 2》
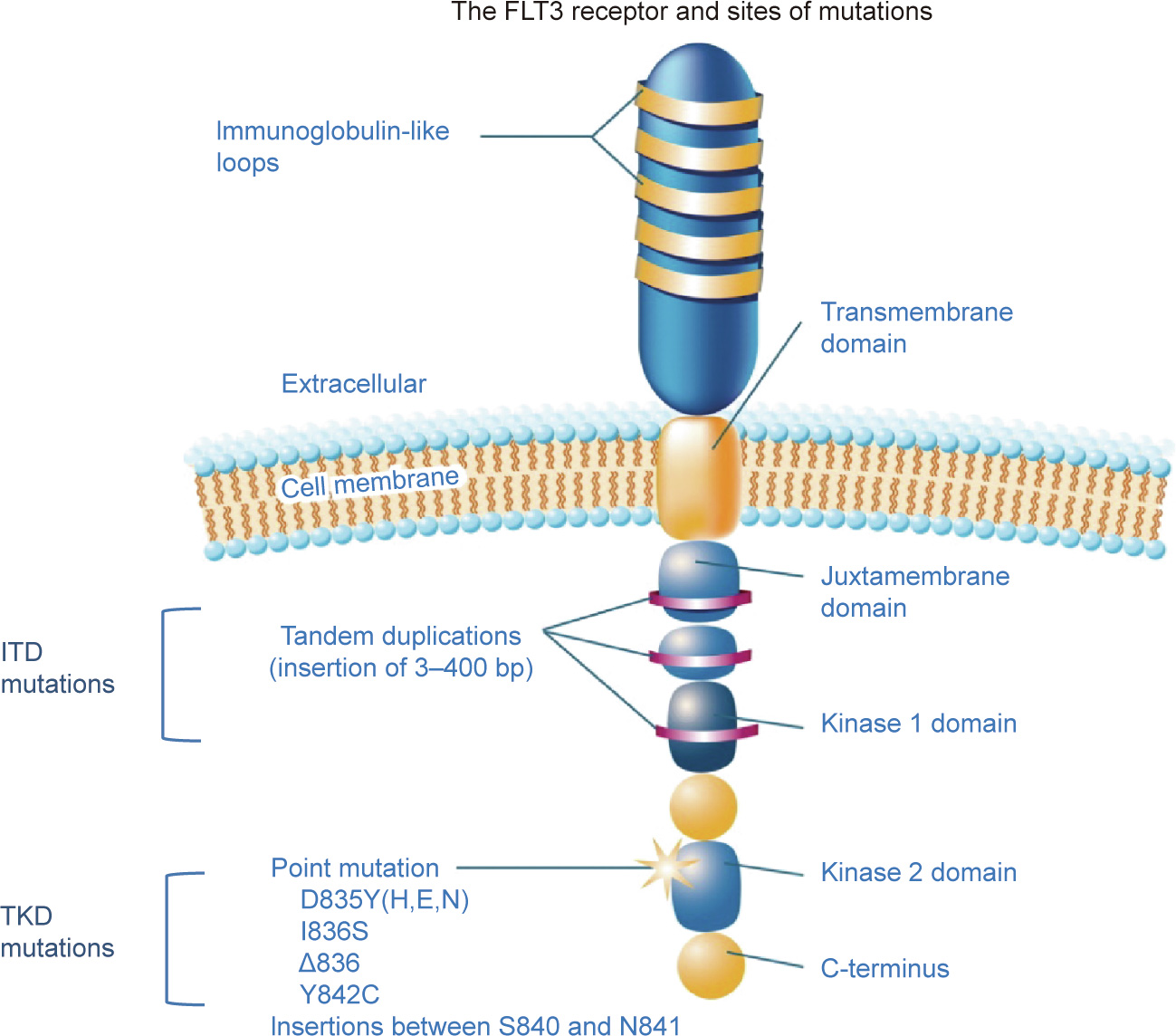
Fig. 2. Schematic depiction of the FLT3 receptor with the most common sites of mutation or alterations indicated. D835Y(H,E,N) indicates the substitution of tyrosine (Y), histidine (H), glutamic (E) acid, or asparagine (N) for aspartic acid at codon 835; I836S indicates the substitution of serine (S) for isoleucine at codon 836; D836 indicates the mutation of three base pairs (bp), affecting codon 836; and the Y842C indicates the substitution of cysteine (C) for tyrosine at codon 842. Reproduced from Ref. [25] with permission of John Wiley and Sons, © 2011.
Routine testing for the FLT3-ITD mutation upon diagnosis is strongly recommended [29] and has now become standard practice. Clonal evolution is an important concept in AML, and it is understood that new mutations can occur during the course of the disease. The FLT3 mutations are no different and have also been shown to appear upon relapse, suggesting the necessity of repeat testing [18]. Moreover, the significant increase of FLT3-ITD mutations that is seen in relapsed AML suggests that the mutation confers a selective advantage to the clone, either directly or indirectly [18]. While evidence has accumulated regarding the prognostic importance of FLT3-ITD in AML, interest has been sparked regarding its therapeutic potential [30]. ITD mutations in FLT3 have been validated in studies as a therapeutic target in the treatment of AML [31].
It is important to realize that the detection of the FLT3-ITD mutation must be interpreted in context. As mentioned earlier, several studies have reported that a high allelic burden confers a higher risk of relapse [26,32,33] as well as inferior long-term survival (Fig. 3). The issue of what constitutes a high allelic burden is far from settled yet, as a large study by the Medical Research Council in the United Kingdom failed to identify a specific allelic ratio cutoff associated with a risk of relapse [34,35]. The length of the FLT3-ITD mutation [36,37] and the insertion site [38] may also have prognostic significance. In the past decade, the interaction between genes and mutations has been emphasized as crucial in the prognostication of AML [39]. Thus, the importance of the mutational burden of the FLT3-ITD mutation cannot be separated from other concurring mutations. Convincing data from several groups demonstrate that the relatively favorable prognosis of low-allelic-ratio FLT3-ITD is only observed in the presence of mutated nucleophosmin (NPM) protein, encoded as the NPM1 gene (Fig. 4) [40,41]. This finding has been widely accepted, as in the European Leukemia Net prognostic classification of AML (Fig. 5) [29]. Due to the relative frequency of the co-occurrence of FLT3- ITD and NPM1 mutations [42] (Fig. 6), this finding has assumed practical significance in the management of AML and has a particular impact on the post-remission strategy [29].
《Fig. 3》

Fig. 3. The results of the UK Medical Research Council (MRC) AML 10 study clearly show the impact of the FLT3 allelic burden on the long-term survival of AML. Green and blue lines correspond to the presence or absence of the nucleophosmin (NPM) encoding gene NPM1 mutation, respectively. CR1: first complete remission. FLT3+low and FLT3+high are defined as ‘‘low-allelic-ratio” (less than 50%) and ‘‘high-allelic-ratio” (more than 50%), respectively. Reproduced from Ref. [33] with permission of Elsevier Ltd., ©2008.
《Fig. 4》

Fig. 4. The interaction of FLT3-ITD mutations and NPM1 in post-remission therapy of AML. (a) In NPM1+ and FLT3-ITDlow, there is no difference in overall survival between chemotherapy and allogeneic transplant. (b) In some populations, if NPM1 is negative, the overall results are significantly worse for both transplant and chemotherapy. (c) In FLT3-ITDhigh, irrespective of NPM1 status, there are dismal results with chemotherapy. Allo HCT: allogeneic hematopoietic cell transplantation. SAL: Study Alliance Leukemia. Reproduced from Ref. [40] with permission of Elsevier Ltd., © 2016.
《Fig. 5》

Fig. 5. Prognostic stratification of AML by genetics. CEBPA: CCAAT enhancer binding protein alpha; MLL: mixed lineage leukemia; inv: inversion; abn: abnormal. Data from an expert panel of the European Leukemia Net (ELN); note that FLT3-ITDlow with NPM1 mutation is considered to be ‘‘favorable” [29].
《Fig. 6》
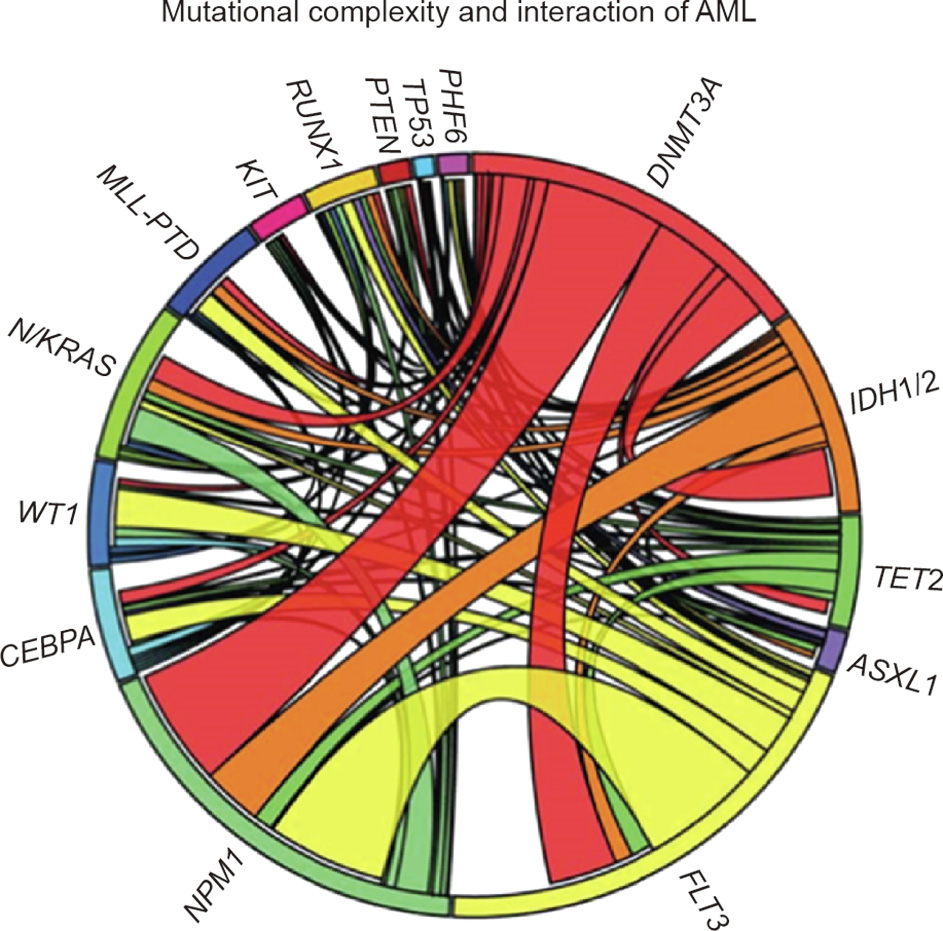
Fig. 6. A Circos diagram depicting the relative frequency and pairwise cooccurrence of mutations in patients with newly diagnosed AML enrolled in the ECOG E1900 clinical trial. The length of the arc corresponds to the frequency of mutations in the first gene, and the width of the ribbon corresponds to the percentage of patients who also had mutation in the second gene. In this case, the two commonest mutations in AML were FLT3, occurring in 37% of patients, and NPM1, occurring in 29% of patients, with a significant group of patients harboring both mutations. Reproduced from Ref. [42] with permission of Massachusetts Medical Society, © 2012.
Once the prognostic impact of FLT3 in AML was established, the development of TKIs blocking FLT3 was a rational therapeutic concept. FLT3 inhibitors can be classified in several ways. First, they are referred to as being either first or second generation. The first generation of FLT3 inhibitors includes midostaurin, sunitinib, sorafenib, lestaurtinib, and ponatinib, while the second-generation inhibitors are crenolanib, quizartinib, and gilteritinib. For the most part, the first-generation inhibitors are considered to be less specific, with multiple targets and a broader spectrum of activity, than the second-generation ones, but that is not always the case [18–20]. It is assumed that first-generation inhibitors have a higher toxicity profile due to their broader potential targets. Second-generation inhibitors have a lower half-maximal inhibitory concentration (IC50) and are likely to have fewer adverse effects.
FLT3 inhibitors can also be categorized as being a type I or type II TKI, based on the way in which they bind to the TKD region of the FLT3 receptor. Type I inhibitors bind to both the active and the inactive conformations of the kinase, but have a higher affinity for the active adenosine triphosphate (ATP) binding region, whereas type II inhibitors bind only to the inactive form [43]. Type II inhibitors fit into a ‘‘back pocket” adjacent to the ATP binding region, which is only available when the enzyme is in its inactive form. Type II inhibitors are more specific, due to the uniqueness of the inactive conformation among the kinases, compared with a similar protein structure of the active form. The missense TKD mutation mentioned above occurs most commonly on the single amino acid exchange at the activation loop residues aspartate [19,21,24,44]. Molecular analysis has shown that the D835 mutation stabilizes the ‘‘active” conformation of the FLT3. Because type II inhibitors are specific for the ‘‘inactive” conformation, the occurrence of the D835 mutation results in resistance to most type II inhibitors, as will be discussed later in more detail. However, there seems to be clinical variability among the type II inhibitors, suggesting that the resistance is not uniform among D835 mutations. Clinical studies do not always report the existence of D835 mutations, although it has been suggested that in the case of type II inhibitors, the mutation may decrease clinical efficacy and, more specifically, limit response duration. It should also be noted that the D835 mutation is becoming recognized as an acquired resistance mechanism, which may be related to prior FLT3 inhibitor administration.
An additional consideration when reviewing the different FLT3 inhibitors pertains to the point in the therapeutic process at which the inhibitor is presumed to be effective. As will be further elucidated, some of the FLT3 inhibitors are being examined for their role in newly diagnosed AML patients, while others are being considered for use in relapsed disease or maintenance therapy. There is also specific discussion of their use following allo-SCT.
《3. Induction therapy》
3. Induction therapy
The story of FLT3’s first known inhibitor, midostaurin, began with the still-ongoing quest to develop selective protein kinase C (PKC) inhibitors [30]. Midostaurin (originally referred to as CGP41251 or PKC412) was derived from staurosporine, a potent inhibitor of PKC activity [45,46]. While the compound did indeed exhibit some activity against PKC, it was soon discovered that the N-benzoyl-staurosporine derivative also inhibited several other important protein kinases. Thus, ensuing studies showed that it was also a potent inhibitor of vascular endothelial growth factor receptor kinase, making midostaurin capable of angiogenesis inhibition. Further investigation demonstrated that midostaurin has inhibitory effects on mutant forms of FLT3 in AML [47] as well as KIT proto-oncogene receptor kinase (KIT) mutations in advanced systemic mastocytosis (SM). It soon became clear that midostaurin is a multi-kinase inhibitor with a broad kinase inhibition profile and with particular significance for hematological malignancies, as it inhibits several important kinases in leukemogenesis [48]. The implications for SM patients with activating KIT D816 mutations have been shown to be clinically relevant, and midostaurin, as monotherapy, has been established as the first line of therapy for this population [49].
Regarding AML with FLT3 mutations, evidence first from in vitro and animal model studies, and then from preclinical and earlyphase clinical trials, clearly showed that midostaurin exhibits inhibitory activity against both the FLT3-ITD and FLT3-TKD mutants [50]. Midostaurin seems to decrease FLT3 autophosphorylation and antagonize downstream signaling. In studies comparing the molecule’s activity on wild-type FLT3 and mutant FLT3, midostaurin demonstrated ten-fold higher selective inhibition of the FLT3 mutant forms (both ITD and TKD) compared with the wild type. Two major metabolites have been identified, both of which exhibit inhibitory activity on FLT3 as well. The compound is orally administered, rapidly absorbed, and generally well tolerated, with the most prevalent adverse effects being gastrointestinal (GI) and hematological, and both being considered mild.
Preclinical studies in FLT3 mutant relapsed/refractory (R/R) AML patients have clearly demonstrated the biological activity of midostaurin in reducing blood and marrow blasts. Notably, there was some degree—albeit to a lesser extent—of blast reduction among non-mutated patients as well. However, it was soon apparent that midostaurin was not adequate as a single agent, and CRs were rare across all study groups [51,52]. Rather than continue the investigation of midostaurin’s potential role in R/R disease, researchers formed a hypothesis suggesting that, for such a nonspecific inhibitor, the best setting to test such a drug’s efficiency is in a newly diagnosed patient, in order to avoid the evolution of resistance mechanisms. Thus, investigators set out to test the notion of improving midostaurin’s demonstrated biological activity by combining it with standard cytotoxic chemotherapy, as a firstline treatment.
The study of midostaurin in AML represents a milestone in the conceptual development of new drugs for AML (Fig. 7). Previously, and for several decades, the prevailing concept was to test a drug at an advanced stage of the disease, usually relapse or refractory. When efficacy was demonstrated at such an advanced stage of the disease, the drug was usually brought forward until, ultimately, it was tested as a first-line therapy. The development of midostaurin involved a key strategic rethinking based on the premise that if a drug shows some activity, such as a reduction of blast cells in the bone marrow, then the lack of a meaningful clinical response should not preclude drug development, nor lead to the conclusion that the drug is not active. Many active drugs have been ‘‘buried” by a failure to elicit a clinical effect when tested at an advanced stage of the disease, when multiple resistance mechanisms are in place. Fortunately, in this case, this unprecedented strategic thinking, which required international agreement, postulated that if there was any biological activity in any drug in the advanced stage, such an agent (given its apparent low toxicity) might well form a cornerstone for adjuvant therapy for patients who have adverse prognostic factors in AML, such as FLT3 mutations. This historic realignment of thinking has now been moved forward to several areas of development, including other drugs for acute leukemia and other diseases.
The Cancer and Leukemia Group B (CALGB) 10603/RATIFY study [53] was an international, randomized, placebo-controlled phase III trial carried out across 17 countries in 225 sites that set out to examine the addition of midostaurin to standard induction and consolidation therapy in patients with FLT3-mutated AML. This landmark study set the proof of concept for the incorporation of FLT3 inhibitors into the standard of care in newly diagnosed patients with AML (Fig. 7(a)). Over the course of three years with extensive academic, government, and industry collaboration, over 3000 newly diagnosed adult patients under the age of 60 were screened for the FLT3 mutation ITD or TKD. Of these, 717 eligible patients were identified and underwent randomization. Patients were stratified according to the type of FLT3 mutation identified: FLT3-TKD (considered a good prognosis), FLT3-ITD with a high allelic-to-wild-type ratio (considered a poor prognosis), and FLT3- ITD with a low allelic ratio (the prognostic significance of this subtype is still undetermined). The primary endpoint was OS, not censored for transplant. The midostaurin group received standard 7 + 3 induction (daunorubicin and cytarabine) and consolidation therapy (high-dose cytarabine), as well as 12 months of maintenance with midostaurin. The placebo group received the same standard induction and consolidation, as well as maintenance with a placebo (Fig. 8).
《Fig. 7》

Fig. 7. Stages in the development of an FLT3 inhibitor as an adjunct to the standard therapy of patients with newly diagnosed AML with mutated FLT3. (a) RATIFY study (using midostaurin); (b) studies comparing midostaurin with other inhibitors; (c) an ideal study would use an FLT3 inhibitor with a separate placebo-controlled randomization at each step of the therapy of AML. MRD: minimal residual disease. ®: randomization.
《Fig. 8》

Fig. 8. Outline of the RATIFY study. DNR: daunorubicin; CR: complete remission; HiDAC: high-dose cytarabine.
The results of the study demonstrated a significantly longer OS in the midostaurin group compared with the placebo group (hazard ratio (HR) = 0.78, p = 0.009). Event-free survival and diseasefree survival were also significantly improved in the midostaurin group across all subtypes (Fig. 9). The rate of CR did not differ between the two groups (Fig. 10), although—importantly—it should be noted that no data are available on the achievement of minimal residual disease (MRD) in either group. The benefit of midostaurin was seen in all subtypes of FLT3 mutation, and there was no difference in severe adverse events between the groups.
《Fig. 9》
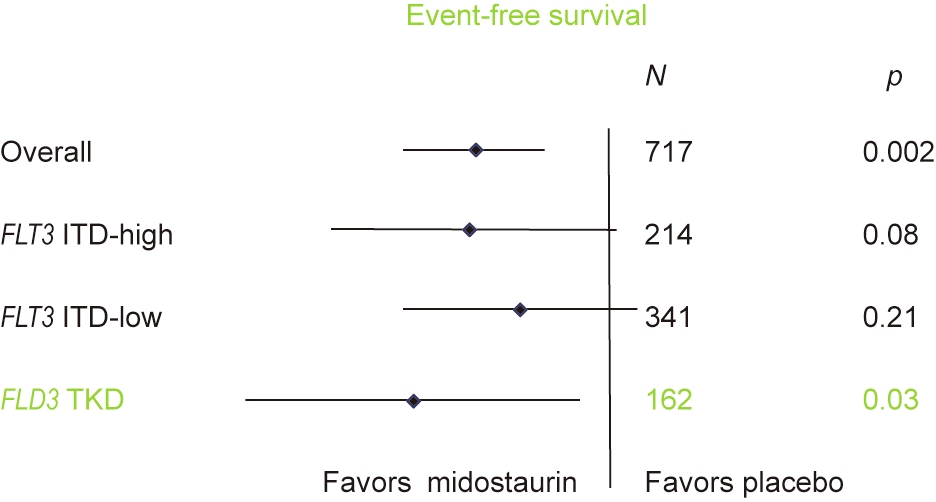
Fig. 9. Improved event-free survival in the RATIFY study for midostaurin across all groups—as seen in this forest plot.
《Fig. 10》

Fig. 10. No difference was found in complete remission rate in the RATIFY study between the midostaurin and placebo groups.
Shortly after this monumental study published its results, midostaurin was approved by the US Food and Drug Administration (FDA) for the first-line treatment of adult patients with FLT3-mutated AML, as an adjunct to the standard 7 + 3 induction regimen of cytarabine and daunorubicin [50]; thus, it became the first targeted therapy to be used in AML.
The results of the RATIFY study have been deemed to be pivotal; some scholars refer to the study as a paradigm shift, as it led to the emergence of the first targeted therapy for AML. Several points should be mentioned. First, the finding that all subtypes seemed to benefit from midostaurin, including the FLT3-TKD subtype that is considered to be more benign, raises a question regarding midostaurin’s mechanism of action. Early clinical trials had demonstrated clear biological activity of the compound against non-mutant FLT3 AML, suggesting that at least some of its effect might be due to the inhibition of other kinases, as well as inhibition of the FTL3 mutation. Indeed, there is an ongoing study by the Spanish PETHEMA group evaluating the type II FLT3 inhibitor quizartinib in AML with wild-type FLT3, and a similar study is using midostaurin (ClinicalTrials.gov NCT 04107727 and NCT 0351297). It is also important to note that the survival curves separate early on in the treatment and remain primarily parallel thereafter, suggesting that most of the therapeutic benefit is achieved at the beginning of treatment. Interestingly, pharmacokinetic studies show that the drug levels are highest during the first few weeks of treatment. An additional finding from subsequent data analysis suggests that, in the RATIFY study, patients who underwent allogeneic transplant during the first complete remission (CR1) benefited more from midostaurin then those who were not transplanted.
The US FDA has not approved the use of midostaurin for maintenance beyond induction and consolidation, although it is approved in Europe for single-agent maintenance as well [54]. In the original RATIFY study, maintenance in the midostaurin group was designed to last for 12 months, but over half the patients (somewhat more than expected) went on to allogeneic transplant upon first remission, and therefore, per protocol, did not receive maintenance therapy. A post hoc analysis of the RATIFY study showed that the study was not able to demonstrate any benefit from maintenance treatment in terms of overall outcomes [55]. Further studies on this issue are pending [56].
Following the publication of the RATIFY study, large phase III randomized studies are now precisely comparing midostaurin, as used in the RATIFY study, with two of the more specific FLT3 inhibitors, gilteritinib and crenolanib (NCT 04027309 and NCT 03258931, respectively) (Fig. 7(b)).
Given the absence of MRD data in the RATIFY study, a fundamental unresolved issue is the precise point in the treatment of AML at which FLT3 inhibitors are most crucial. If an inhibitor is active in induction, how important is further exposure in consolidation and, more importantly, in maintenance therapy? While some ongoing randomized studies are investigating maintenance therapy (e.g., a placebo-controlled study of gilteritinib, NCT 02927262), an ideal study would use an FLT3 inhibitor with a separate placebo-controlled randomization at each step of the therapy of AML: induction, consolidation, and maintenance (Fig. 7(c)). Although such a clinical trial would have been ideal at an earlier phase of development, given the current widespread use of and belief in FLT3 inhibition throughout AML therapy, such a study is unlikely to ever be conducted.
《4. Relapsed/refractory AML》
4. Relapsed/refractory AML
《4.1. Gilteritinib》
4.1. Gilteritinib
Gilteritinib (ASP2215, Xospata) is an inhibitor of AXL, a smallmolecule tyrosine kinase receptor that is crucial for the growth of FLT3-ITD in AML [57]. The ADMIRAL study was a pivotal trial evaluating the use of gilteritinib, in comparison with standard chemotherapy, for the treatment of advanced FLT3-mutant AML. The data firmly placed gilteritinib in the forefront of therapy for R/R FLT3-mutant AML [58].
This large international phase III trial enrolled adults over 18 years of age who either had relapsed AML following a CR to standard therapy or were refractory to induction therapy. Patients with either FLT3-ITD or TKD mutations were randomized to receive either gilteritinib as monotherapy or any of the standard regimens for salvage chemotherapy, preselected in advance by the investigators. Approximately one-fifth of the patients in both groups underwent a previous allo-SCT. The primary endpoints were OS and the rate of achievement of a full or composite CR. The study reported a superior OS in the gilteritinib arm (9.3 vs 5.6 months; p < 0.001). The conclusion of the study was that gilteritinib led to higher response rates and longer survival time than salvage chemotherapy. Gilteritinib monotherapy is now approved for R/R FLT3- mutated AML in Europe and in the United States [18]. An updated analysis of the data reported that more patients receiving gilteritinib achieved CR or a composite CR, and more patients were able to proceed to allo-SCT [58].
The ADMIRAL study, described in detail here, represents another milestone in the development of FLT3 inhibitors for AML. Coming after the early failures of lestaurtinib, sorafenib, sunitinib, and midostaurin [51,59–61] to achieve a meaningful clinical response in advanced disease, it took a courageous effort to embark on a phase III study with a second-generation FLT3 inhibitor as monotherapy. While the use of the drug as a single agent was necessary in order to unequivocally demonstrate its activity and thus gain regulatory approval (Fig. 11(a)), it is likely to be used in practice as an adjunct to other potent chemotherapy regimens, at least in young adults. Such clinical trials are likely to be conducted soon (Fig. 11(b)), particularly in countries where the off-label use of drugs is prohibited. It should be noted that a large randomized trial of chemotherapy followed by lestaurtinib for FLT3-mutant AML in first relapse did not show an increased response rate. However, in this trial, only a small percentage achieved sustained FLT3 inhibition [62].
《Fig. 11》

Fig. 11. Stages in the development of second-generation FLT3 inhibitors in R/R AML. (a) The use of the drug as a single agent; (b) use of drug as an adjunct to other potent chemotherapy regimens.
The results of the ADMIRAL study offer other intriguing possibilities. If gilteritinib as monotherapy improves survival in advanced disease, is it not likely to be efficacious in de novo AML as well? Of particular interest would be the clinical scenario of an older adult with FLT3-mutated AML, who is unfit to receive intensive chemotherapy. Should gilteritinib, with its low toxicity, be the preferred option, rather than venetoclax-based therapy, or even be considered as an adjunct to such therapy? Such studies are likely to be conducted in the near future.
《4.2. Crenolanib and quizartinib》
4.2. Crenolanib and quizartinib
Crenolanib exhibits inhibitory activity against both FLT3-ITD and FLT3-TKD. It also shows inhibitory action against the PDGFR. Encouraging data for the use of crenolanib in FLT3-mutant AML was obtained from several phase II studies in patients with R/R disease. A small study evaluating crenolanib in 38 patients reported a CR with incomplete hematologic recovery (CRi) of 23% among patients who were naïve to FLT3 inhibitors [63]. A second, larger study of 69 patients in a similar patient population reported a CRi of 39%. Of particular biological and clinical interest was a significant response seen also in patients who had received prior therapy with FLT3 inhibitors [64]. The drug was not approved by the US FDA but was approved by the European Medicines Agency (EMA). A phase III study of crenolanib versus midostaurin following induction and consolidation therapy in newly diagnosed FLT3-mutated AML is ongoing (NCT 02668653). If the results are positive, this study may lead to a further improvement in the therapy of newly diagnosed AML with FLT3.
Quizartinib is a potent and selective inhibitor of FLT3-ITD, but not of FLT3-TKD [65–67]. Several studies, both phase I and phase II, of quizartinib in R/R AML reported a response rate of greater than 50% in AML with mutant FLT3-ITD [68]. QT-interval (from the beginning of the QRS complex to the end of the T wave) prolongation was a noticeable toxicity. One of the major studies with quizartinib was the QuANTUM-R study, a randomized phase III trial conducted in 19 countries among adults aged 18 years or older with R/R FLT3-ITD AML following standard therapy. Patients were randomly assigned to quizartinib or to the investigator’s choice of preselected chemotherapy, as in the ADMIRAL study. Of the patients in the quizartinib group, 23% had undergone alloSCT as a previous therapy, compared with 22% in the chemotherapy group. The primary endpoint was OS. A total of 367 patients were enrolled, 245 of whom were randomly allocated to quizartinib and 122 to chemotherapy. The median OS was 6.2 months in the quizartinib group and 4.7 months in the chemotherapy group (p = 0.02). Apart from QT prolongation in the quizartinib arm, the major toxicities in both groups were those that are typically observed after myelosuppressive therapy. The percentage of deaths was 33% in the quizartinib group and 17% in the chemotherapy group. Quizartinib clearly presents a novel option for the therapy of advanced FLT3-mutated AML. Its selectivity for FLT3-ITD is attractive and accounts for the potent efficacy observed in all the early studies [68].
Even though the results from the study indicated an improvement in OS for the patients who were given quizartinib, the study had important limitations due to adverse events, so the overall effectiveness of quizartinib could not be convincingly demonstrated. As a result, the EMA declined marketing authorization, as did the US FDA following a recommendation from the organization’s Oncologic Drugs Advisory Committee (ODAC). Before the ODAC meeting, the US FDA conducted its own efficacy analysis and determined that the median OS was 26.9 weeks with quizartinib compared with 20.4 weeks with chemotherapy (p = 0.019). Although this analysis confirmed an advantage for quizartinib, several questions persisted. Among other issues, these questions concerned the number of patients that had been randomized but not treated and the heterogeneity in the follow-up therapies that patients received after discontinuing study treatment, both of which confounded the assessment of the OS endpoints. These rulings have been questioned by many clinicians, given the general similarity of the QuANTUM-R and the ADMIRAL studies, both of which were investigated in a difficult patient population where there is no standard of care for R/R AML. In contrast, in Japan, the Ministry of Health, Labor, and Welfare (MHLW) reviewed the data of the QuANTUM-R study and approved the use of quizartinib for the treatment of adult patients with R/R FTL3-ITD-positive AML.
《5. Other FLT3 inhibitors》
5. Other FLT3 inhibitors
《5.1. Sorafenib》
5.1. Sorafenib
An additional multikinase inhibitor that deserves consideration in first-line AML therapy is sorafenib. As was the case with midostaurin, sorafenib was not originally designed to block the FLT3. This compound has been shown to inhibit several kinases that play a part in cell proliferation and division as well as in leukemogenesis, such as RAS/RAF, KIT, platelet-derived growth factor (PDGF) and vascular endothelial growth factor (VEGF) receptor, and FLT3. Sorafenib is currently approved for the treatment of renal cell carcinoma and hepatocellular carcinoma [69]. Its efficacy in AML is feasible via its potential inhibition of FLT3 mutations, but may also be due to its proven interference with the RAS/RAF signaling cascade [70]. As early as 2004, sorafenib demonstrated inhibitory in vitro activity against FLT3-mutant human and murine cell lines, and clinical trials in AML patients began [71,72].
One phase I/II trial tested the tolerability and efficacy of combining sorafenib with induction therapy (in this case, cytarabine and idarubicin) [73,74]. The results of that study indicated a high response rate, particularly among FLT3-ITD mutated patients; however, the majority of the patients relapsed in long-term follow up. The drug was generally well tolerated; adverse effects included GI toxicities, infections, and a few cases of hand-and-foot syndrome.
The first randomized placebo-controlled trial with sorafenib, by the Study Alliance Leukemia (SAL) group, was conducted among newly diagnosed AML patients above the age of 60 [75]. In this study, sorafenib was administered to patients on day 3 after standard 7 + 3 induction, and continued to be administered until three days before the next chemotherapy course. This study not only demonstrated more adverse effects among patients receiving sorafenib, but also failed to show therapeutic benefit for this treatment. However, it was suggested that younger patients without comorbidities may better tolerate the additional medication. Therefore, the SORAML trial was designed, enrolling only previously healthy adult AML patients younger than 60 and including also those without FLT3 mutations [76]. This was a randomized, double-blind, placebo-controlled phase II trial among 276 patients across Germany. With this younger population, the results did, in fact, demonstrate that the addition of sorafenib to standard induction therapy provided therapeutic benefit. The primary endpoint of the study was the median event-free survival (EFS) (Fig. 12 [76]), which was 40% in the sorafenib arm versus 22% in the placebo group after three years (HR = 0.65, p = 0.012). There was also a significant prolongation of relapse-free survival (RFS) in the sorafenib group. The OS was not significantly different between the groups, possibly due to the relatively small number of patients in the study, which was not powered for OS. In an important five-year follow-up analysis [77] the difference in OS was more pronounced, with 8% higher OS in the sorafenib group, although the difference did not reach statistical significance. An exploratory analysis of the 46 patients with FLT3-ITD mutations reported an improvement in OS and RFS in the sorafenib group versus the placebo group, although this was not statistically significant. It is interesting to note that the EFS remained significant even after removing the FLT3-mutated patients from the analysis, suggesting that sorafenib has some broad activity in non-mutated patients as well.
A recent double-blind study by the Australian Leukemia and Lymphoma Group (ALLG) of sorafenib versus a placebo, in combination with standard chemotherapy, failed to show a significant improvement in either EFS or OS [78].
《Fig. 12》

Fig. 12. The SORAML study compared sorafenib with a placebo in newly diagnosed patients with AML. Reproduced from Ref. [76] with permission of Elsevier Ltd., ©2015.
《5.2. Sunitinib》
5.2. Sunitinib
Sunitinib is another example of a small molecule that was first recognized as a potent inhibitor of the PDGFRs, VEGFRs, and KIT receptors. It was then characterized as an FLT3 inhibitor, and was shown to reduce the phosphorylation of wild-type and mutant variants of the kinase [79]. A phase I trial showed modest singleagent antileukemic activity [61]. Subsequently, a phase I/II trial investigated the combination of sunitinib with standard induction therapy in newly diagnosed AML patients [80]. In this study, 50% of FLT3-ITD mutated patients and 38% of the FLT3-TKD patients achieved CR. The reported toxicities are GI toxicity, mucositis, and fatigue. This agent is not currently being investigated for the treatment of AML.
《5.3. Lestaurtinib》
5.3. Lestaurtinib
Lestaurtinib is an additional FLT3 inhibitor that has been investigated for first-line AML patients. Like midostaurin, sorafenib, and sunitinib, lestaurtinib is a first-generation FLT3 inhibitor [81,82]. This drug was tested as part of the AML15 and AML17 studies, which were large, prospective phase III multicenter trials for patients with either myelodysplastic syndrome (MDS) or newly diagnosed AML. A total of 500 patients with FLT3 mutations were randomly assigned to receive lestaurtinib in addition to standard induction or induction alone. The trials ran consecutively, and the data was meta-analyzed. The study showed that the drug was well tolerated but failed to show an overall clinical benefit. Lestaurtinib was also evaluated as a first-line treatment for AML patients who were deemed unfit to undergo standard intensive chemotherapy, primarily due to age [83]. In a phase II trial, lestaurtinib was administered as monotherapy for eight weeks. The results showed only modest clinical efficacy, and there were no CRs [59]. In another randomized study for patients with relapsed AML, no benefit was demonstrated for lestaurtinib when it was added following salvage chemotherapy [62].
《6. Post-transplant maintenance therapy》
6. Post-transplant maintenance therapy
Following successful induction in AML, allogeneic transplant is generally considered to be common practice in CR1 in all patients except those with a favorable karyotype. In FLT3-ITD-mutated patients in particular, studies have shown improved disease-free survival after transplant [84,85]. The concept of maintenance following transplant, however, is quite controversial in all types of AML [86,87]. Not only in AML, but also in other hematologic diseases, the evidence for maintenance therapy following transplant is conflicting, with the exception of TKIs for Philadelphia chromosome-positive ALL, if positive for MRD [88,89]. Considering the immunosuppressive state of such patients, it is not always clear whether the risks of maintenance treatment outweigh the benefits. On the other hand, the post-transplant setting is considered to be unique, in that the tumor burden is assumed to be at its lowest. Moreover, it is theorized that the newly transplanted, non-exhausted immune system may have the ability to better optimize antileukemia agents than in other settings [90]. With the advent of specific FLT3-ITD inhibitors, the debate about maintenance following transplant is being rekindled. The fact that the FLT3-ITD clone has been shown to be a dominant one in relapse seems to emphasize the potential of targeting this mutation in maintenance.
《6.1. Sorafenib》
6.1. Sorafenib
The first FLT3-ITD inhibitor to be investigated in this context was sorafenib. Although not the TKI of choice for most AML studies, its development was uniquely pursued, including randomized studies, and will therefore be discussed here in some detail. In 2014, a phase I trial enrolled 22 FLT3-ITD mutated patients in a dose-escalation cohort design, aimed at defining the maximum tolerated dose of the drug in post-allogeneic transplanted patients[91]. The only patients included were those who had complete morphological remission, who had at least 70% chimerism of donor origin, and who had a recovered platelet and neutrophil count. Sorafenib was administered anywhere between 45 and 120 days following the transplant, allowing for variability in the patients’ recovery time. It was given on a daily basis in 28-day cycles. The results of this study indicated that sorafenib was safely tolerated in the post-transplant setting, with a maximally tolerated dose ranging between 200 and 400 mg twice daily. The toxicities were primarily GI and skin related, as has been reported in other studies. Some patients experienced erythematous skin rashes that resembled acute graft-versus-host disease (GVHD) but were resolved upon discontinuation of the drug. The results of the study seemed to indicate that sorafenib offered an advantage in overall and progression-free survival. The same group then followed up with a retrospective study [92]. In that study, 81 AML patients with the FLT3-ITD mutation were identified, all of whom had undergone allogeneic transplant while in CR1 after induction therapy. Of these patients, 26 comprised the sorafenib group and received posttransplant sorafenib maintenance therapy. The control group consisted of similar patients who did not receive sorafenib following transplant. The results of the study showed an improved twoyear OS in the sorafenib group (81% vs 62%), as well as an improved two-year progression free survival (PFS) (82% vs 53%). There was also a lower two-year relapse incidence (8.2% vs 37.7%). The dosing ranged from 200 to 400 mg, with reductions occurring as needed. All but two patients had to discontinue sorafenib at some point, but most were able to tolerate sustained treatment for a median of 336 days. An additional retrospective study examined 27 AML patients with FLT3 mutations (two of which were TKD mutations, while the remaining were ITD) who received sorafenib posttransplant [93]. The one-year OS and progression-free survival were both 92%; most toxicities were mild and all were reversible, responding for the most part to dose reduction. Several other studies reported similar results [94,95].
Following the promising results of retrospective studies, a number of prospective trials were launched. Pratz and colleagues [90] enrolled 44 patients who underwent allo-SCT and tested positive for FLT3-ITD mutations. Sorafenib was administered both post-transplant and pre-transplant, following induction/consolidation. The patients began with 200 mg twice daily, with dose escalation after seven days, and dose reductions whenever grade 3 or 4 toxicity occurred. Elevated hepatic enzymes were the most common grade 3 or 4 toxicities, with thrombocytopenia being almost as common. Most patients were unable to tolerate the 400 mg dosage. Blood samples throughout the study analyzed FLT3 inhibition in vitro, expressed as the percentage of baseline FLT3 phosphorylation. Notably, FLT3 inhibition seemed to correlate with the tolerability-determined dosing, suggesting that when the dosage was reduced due to toxicity, the FLT3 inhibition remained. The OS was 76% at 36 months, but the EFS was only 64%. The possibility that GVHD may actually increase with sorafenib treatment is interesting, as it suggests that this may contribute to the therapeutic effect. It has been theorized, based on animal studies, that sorafenib may enhance the graft-versus-leukemia effect without increasing systemic GVHD, by directly inducing interleukin (IL)-15 production in leukemia cells [96].
An additional study to consider is the SORMAIN study [97]: a randomized, double-blind placebo-controlled phase II study that took place in Germany and Austria among AML FLT3-ITDmutated patients, all of whom had undergone allogeneic stem cell transplantation. The patients were randomized to receive either a placebo or sorafenib for up to 24 months, beginning between day +30 to day +100 following transplant. The initial results were quite promising, with a two-year RFS of 85% in the sorafenib maintenance group versus 53.3% in the placebo-treated group. However, the study was terminated early due to slow accrual, with only 83 patients.
A recent study from China aimed to validate these results [98] in an open, randomized phase III trial. A total of 202 newly diagnosed AML patients with FLT3-ITD mutations were allocated. All patients received their allogeneic transplant in CR1 and had hematopoietic recovery within 60 days of transplant. The patients were randomized into either the control group (N = 102) or the sorafenib group (N = 100), in which they received sorafenib starting on day 30–60 until day 180 post-transplant. The initial dose was 400 mg twice daily. Among the patients who received sorafenib for maintenance after transplant, there was only a 7% one-year relapse, compared with a 24.5% relapse among the controls. The relapse incidence at two years also indicated an advantage for the sorafenib group, with an 11.9% relapse versus 31.6% among the controls. OS and leukemia-free survival were also improved in the sorafenib group. There were no treatment-related deaths, and the sorafenib was well tolerated. An independent study committee was tasked with determining whether abnormal symptoms (particularly skin-related symptoms), signs, or laboratory results were to be attributed to GVHD or to sorafenib treatment. The overall adverse events were similar between the study group and the control. Approximately 60% of the patients receiving sorafenib needed dose reductions due to toxicity, with the most common adverse events being hematological, skin related, and GI. Similar toxicities have also been reported by others [99]. This study was limited first and foremost because it was not blinded; however, apart from demonstrating the tolerability of post-transplant sorafenib maintenance, it does seem to indicate that efficacy is likely.
In a recent comprehensive review, the European Society for Blood and Marrow Transplantation (EBMT) Acute Leukemia Working Party published clinical practice recommendations on transplant in AML patients with FLT3-ITD mutations [100]. They unequivocally advocate post-transplant maintenance with an FLT3 inhibitor. Based on the aforementioned studies, they suggest that sorafenib be used as the current preferred agent, pending prospective clinical trials evaluating other FLT3 inhibitors in the post-remission setting. In practice, based on the existing data, sorafenib appears to be the most commonly used TKI for posttransplant maintenance.
《6.2. Midostaurin》
6.2. Midostaurin
Midostaurin is also being evaluated for a post-transplant role. A randomized, unblinded, phase II randomized trial enrolled 60 FLT3- ITD-mutated patients in a study called RADIUS [101]. In this study, half the patients were randomized to receive midostaurin anywhere from 28 to 60 days post allo-SCT. This treatment continued for 12 months, and the patients were followed up for 24 months. The results demonstrated a 76% leukemia-free survival at 18 months for the control group, compared with 89% in the group randomly allocated to receive midostaurin maintenance—a difference that was not statistically significant. An additional study evaluated midostaurin in this setting in a prospective, phase II multiinstitutional trial [56]. In this study, 284 patients with FLT3-ITD mutations were enrolled, all of whom received standard 7 + 3 therapy in addition to oral midostaurin for induction. Patients eligible for allo-SCT continued on to transplant, which was followed by midostaurin maintenance from day 30 post-transplant, if possible, until no later than 100 days after. Those who were unable to undergo transplant received midostaurin until after the last highdose cytarabine HiDAC consolidation cycle. Compared with historical controls, the results of this study showed better EFS and OS for the midostaurin-treated patients. The purpose of this study was also to demonstrate the efficacy of incorporating midostaurin into both the induction and post-transplant maintenance. For older patients, the two-year EFS was 34%, and the OS was 46%. It should be noted that most of the patients prematurely discontinued the drug, primarily due to toxicity.
《6.3. Gilteritinib》
6.3. Gilteritinib
Gilteritinib is a third FLT3 inhibitor that is under consideration for use after transplant. Several authorities suggest that this drug is safer and better tolerated than midostaurin or sorafenib [87], while others consider the toxicity profiles of the latter to be entirely manageable [102]. A phase III, randomized, placebo-controlled, multicenter study comprising 149 sites worldwide, the blood and marrow transplant clinical trials network (BMT-CTN) 1506, is currently underway to evaluate the use of gilteritinib as maintenance therapy after allogeneic transplant in patients with FLT3-ITD positive AML [87,103]. In this study, post-transplant FLT3-ITD AML patients are randomized into either gilteritinib or placebo maintenance post-transplant groups. The study aims to determine the benefit of gilteritinib maintenance in the post-transplant setting and will evaluate MRD both pre- and post-transplant, an aspect that has not yet been investigated in previous studies. With a target accrual of 350 randomized patients, this is a definitive study and the results, expected later in 2021, are awaited. However, the lack of a control arm with sorafenib will make clinical interpretation difficult, and several investigators have emphasized the need for a direct evaluation of gilteritinib versus sorafenib in the post-transplant setting [93,102].
With the integration of FLT3-ITD inhibitors into clinical practice, it is likely that we will see more prospective studies in the future evaluating the best therapy for maintenance post-allogeneic transplantation.
《7. The combination of FLT3 inhibitors with other targeted agents》
7. The combination of FLT3 inhibitors with other targeted agents
The possibility of combining FLT3-ITD inhibitors with other novel agents in AML is a natural development that aligns with the current practice of using targeted agents. Venetoclax is a small molecule that binds to the anti-apoptotic protein Bcl-2 and selectively inhibits its activity [104]. It is used as a single agent in some lymphoid malignancies, but has shown only very limited activity in AML when used alone. However, the now well-known and widespread combination of venetoclax with a hypomethylating agent or low-dose cytarabine is proving to be successful and is the first-line treatment for de novo AML in patients unfit for standard induction therapy [11,105]. Ma et al. [106] examined the in vivo and in vitro effects of combining FLT3 inhibitors with Bcl-2 inhibitors. In their study, they showed that both midostaurin and gilteritinib enhanced the antileukemic effect of venetoclax in FLT3-ITDmutated AML cell lines. In FLT3-ITD-mutated xenograft mice models, they also demonstrated in vivo efficacy of this combination. A number of additional studies have validated and expanded upon these results both in vivo and in vitro [107,108]. A mutational analysis of AML patients treated with venetoclax suggested that FLT3- mutated patients developed resistance faster than those with other mutations [109]. That study, in conjunction with the preclinical data, suggests the possibility of a synergistic relationship between these two drug classes. Several clinical trials currently underway are examining the combination of venetoclax and FLT3 inhibitors in AML patients with the FLT3-ITD mutation. Thus, for example, a recent study from the University of Pennsylvania in Philadelphia, USA, reported encouraging results using gilteritinib with venetoclax [110].
An additional area of interest is the combination of hypomethylating agents such as decitabine or azacitidine with FLT3 inhibitors. In vivo studies, such as those conducted by Chang et al. [111], have suggested that the combination of either decitabine or azacitidine with sorafenib or quizartinib is synergistically cytotoxic for leukemic cells. Several phase I + II trials have examined various combinations, such as midostaurin with azacitidine [112] or with decitabine [113], as well as sorafenib with azacitidine [114] or with decitabine [115]. These promising combinations seem to be well tolerated.
An ongoing trial from the MD Anderson Cancer Center in Houston, Texas, USA, is studying the triple combination of quizartinib, decitabine, and venetoclax in newly diagnosed R/R patients with FLT3-mutated AML. The initial report is clearly encouraging [116].
Several other combinations are currently being investigated. Dayal et al. [117] successfully used a collaborative dual FLT3/TOPK inhibitor, HS1169, to act against FLT3-ITD and sorafenib-resistant cell lines. Furthermore, an autophagy inhibitor, TAK-165, can induce cancer cell death through the activation of chaperonemediated autophagy to improve the effectiveness of cancer therapies [118]. By integrating these novel inhibitors with FLT3 inhibitors, their efficacy may be further improved [119].
Immunotherapies are also potential agents for coupling with FLT3 inhibitors. Thus, for example, a phase II study is examining the combination of gilteritinib with the programmed death-ligand 1 (PD-L1) checkpoint inhibitor atezolizumab (NCT 03730012) in R/R patients with FLT3-mutated AML. Furthermore, in the exciting and rapidly evolving domain of chimeric antigen receptor (CAR) T-cell therapy, FLT3 ligand (FL)-based targeting is being considered [120]. Jetani et al. [121] examined the combination of CAR T-cells targeting FLT3-ITD AML together with crenolanib. The reported data suggest a synergistic cytotoxic effect of this combination. It is therefore likely that we will see many such studies in the future, incorporating some of the novel targeted agents in AML.
《8. Resistance mechanisms for FLT3 inhibitors》
8. Resistance mechanisms for FLT3 inhibitors
Virtually all targeted agents in AML can be expected to develop resistance. This is inherent to the biology of AML, where there are multiple driver mutations. FLT3 inhibitors are no different, and various mechanisms for the development of such resistance have been described. It is therefore not surprising that although many studies have reported that FLT3 inhibitors have favorable clinical activities for AML patients with FLT3, the response duration remains short due to the rapid development of resistance. This is particularly so when FLT3 inhibitors are used as monotherapy, and is the primary rationale for the use of combination therapy. Successful AML treatment has always been hampered by high relapse rates, which are attributed to either primary or acquired resistance. The clonal nature of the disease predisposes it to develop resistant clones, although there are several other mechanisms of resistance development as well [122,123].
The selection pressure applied to the AML clones during induction treatment is an important concept. The theory is that the clones who survive the initial chemotherapy proliferate and cause relapsed disease. In vitro studies have demonstrated that the treatment of FLT3-ITD mutated AML cell lines with FLT3 inhibitors led to the development of FLT3-TKD mutated cells [124]. As mentioned previously, mutations in the TKD region confer resistance to many of the FLT3-inhibitors—particularly type II inhibitors, due to their binding site. Emerging point mutations in the TKD region following FLT3 inhibitor treatment, most commonly at the D835 residue at the activation loop, have been well described. However, there are clearly other mechanisms at play, as only about half of relapsed FLT3-mutated patients have TKD mutations.
Different FLT3 inhibitors have been shown to cause different resistance profiles via different mechanisms. Thus, for example, the mutation at the D835 residue has been described in patients who relapsed on type II inhibitors such as quizartinib [44] and sorafenib [125] therapy, and the TKD N676K mutation has been shown in patients treated with midostaurin [122]. An additional TKD mutation confers resistance to crenolanib but not to ponatinib. Following treatment with some inhibitors such as midostaurin and crenolanib [126], patients have been shown to develop FLT3 independence, whereas the FLT3-ITD mutation was lost completely upon relapse.
An additional resistance mechanism relates to the FL, whose increased concentration in patients’ plasma after induction therapy has been correlated with reduced FLT3-ITD inhibitor activity. It is presumed that the FL acts via the activation of wild-type-FLT3, which re-phosphorylates downstream molecules, particularly the mitogen-activated protein kinase (MAPK) pathway, and thus counteracts the FLT3-ITD inhibition. Upregulation of the FL, or high levels of wild-type-FLT3 to begin with, seem to contribute to the development of FLT3 inhibitor resistance [127].
Another adaptation that may play a part in developing resistance is the upregulation of oncogenic kinases in response to FLT3 inhibitors. It has been shown that kinases that are constitutively activated by FLT3-ITD mutations, such as phosphatidylinositol 3-kinase (PI3K) and MAPK, acquire escape mechanisms in response to FLT3 inhibition, which presumably contribute to resistance. It has even been suggested that the inhibition of some of these kinases during treatment with FLT3 inhibitors may decrease the development of resistance [128].
As the use of FLT3 inhibitors expands, the resistance mechanisms continue to develop, and the need for strategies to overcome them increases. One straightforward notion applied in oncology and in other fields (e.g., anti-microbial treatment) is to combine several agents with different mechanisms of action up front. An additional method, mentioned earlier, is the concurrent inhibition of downstream kinases such as MAPK and signal transducer and activator of transcription 5 (STAT5) during treatment with FLT3 inhibitors. Another point to consider is interclass switching, particularly between type II and type I inhibitors. Thus, for example, crenolanib, a type I inhibitor, is inherently active against D835 mutations, which are generally resistant to type II inhibitors.
《9. Summary and conclusions》
9. Summary and conclusions
Molecular targets have transformed the prognostic and therapeutic landscape in AML. Since the initial discovery in 1996 of the FLT3-activating mutations, rapid advances have occurred, ultimately changing the standard of care in AML. FLT3 inhibitors are routinely incorporated into the therapy of 25%–30% of AML patients who harbor the mutation. The mostly nonmyelosuppressive and generally well-tolerated toxicity profiles of FLT3 inhibitors have led to their common use. While recognizing that the presence of the FLT3-ITD mutation still confers an adverse prognosis, the development of targeted inhibitors has clearly improved the short- and long-term prognosis of such patients, and has led to an increasing number of patients achieving MRD negativity, and possibly reduced the need for more intensive therapies such as allo-SCT. The development of resistance remains an ongoing challenge. All these aspects were considered in this review, which highlighted the dramatic contribution that targeted therapies have made to current practice.
《Acknowledgment》
Acknowledgment
We wish to thank Beth Zisman for assistance in the preparation of this manuscript.
《Compliance with ethics guidelines》
Compliance with ethics guidelines
Meira Yisraeli Salman, Jacob M. Rowe, and Nir Weigert declare that they have no conflict of interest or financial conflicts to disclose.











 京公网安备 11010502051620号
京公网安备 11010502051620号




