

《1. Introduction》
1. Introduction
The current coronavirus pandemic, which has been ongoing since December 2019 (coronavirus disease 2019 (COVID-19)), has become a global health emergency [1–3]. The disease is caused by a newly identified beta-coronavirus, human coronavirus 2019 (HCoV-19) (also known as human coronavirus 2019 (SARS-CoV2)), which is closely related to the SARS-CoV. Like SARS, HCoV-19 causes lower respiratory tract infection (LRTI), which may eventually progress to atypical pneumonia, requiring intensive care and mechanical ventilation [4–6]. Compared with SARS-CoV, HCoV19 is more contagious and has infected far more people worldwide, thus causing substantially more casualties [4,5]. Although possible viral reservoirs including bats and pangolins have been suggested as sources of HCoV-19 [3,7], more investigation is needed to ascertain its source and routes of zoonotic transmission [8–10]. To date, no vaccine or remedy specific for coronaviruses infection—including that of HCoV-19—is available.
Similar to SARS-CoV, the virion surface spike glycoprotein encoded by the HCoV-19 S gene is essential for target cell attachment and fusion processes [11–13]. Given the importance of the spike protein (S protein) in COVID-19 pathogenesis and in its potential immunogenicity for vaccine development, global efforts have been made to elucidate the structure of the S protein, beginning shortly after the first HCoV-19 sequence was published [1,13,14]. The HCoV-19 S glycoprotein is a 1273-amino acid precursor polypeptide; it can be cleaved by host cell proteases (cathepsin L, TMPRSS2) into the S1 fragment, which contains the receptor binding domain (RBD) to attach host receptor human angiotensin I converting enzyme 2 (hACE2), and the S2 fragment, which is responsible for the subsequent membrane fusion [1,12,13]. The S protein is predicted to have a cleavable Nterminal signal sequence (1–15), which presumably directs the protein toward the endoplasmic reticulum (ER) for extensive glycan decoration prior to virion packing.
Glycosylation is one of the most prominent post-translational modifications (PTMs) in many viral spike or envelope proteins, and has been shown to mediate host attachment, immune response, and virion packaging and budding [15–24]. In particular, the binding of coronavirus S proteins to their respective receptors has been shown to be mediated by viral oligomannose N-glycans [19,20,23]. In addition, C-type lectin dendritic cell-specific ICAM3-grabbing non-integrin 1 (DC-SIGN) and liver/lymph nodespecific ICAM-3-grabbing non-integrin (L-SIGN) can enhance viral entry via their binding to the S protein glycans [25,26]. Therefore, numerous antiviral strategies have been designed to interfere with protein glycosylation or glycan-based interaction [21,24,27,28]. Glycosylation is also considered to be a key aspect in the development of an effective vaccine [29,30].
Like the SARS-CoV S protein, which contains 23 N-linked glycosylation sequons (N-X-S/T, X ≠ P), the HCoV-19 S protein is predicted to host 22 sequons per protomer or 66 per trimer [13]. However, the potential glycosite pattern in the S1 of the HCoV19 S protein is different from that of SARS-CoV, while the glycosylation sites in the S2 region are significantly conserved between HCoV-19 and SARS-CoV. Such a difference in the S1 glycosylation pattern may be associated with the fact that the biological and clinical characteristics of HCoV-19 are profoundly different from those of other coronaviruses. In this study, we report a comprehensive N-glycosylation profile—as well as other PTMs—of the HCoV19 S protein and hACE2, elucidated by high-resolution mass spectrometry (MS) analyses. Based on these analyses, we reinterpret the current HCoV-19 S protein structural model to highlight important glycan features related to COVID-19 pathogenesis. Nonetheless, we demonstrate that the binding of the HCoV-19 S protein and hACE2 is independent of their glycosylation status.
《2. Material and methods》
2. Material and methods
《2.1. Expression and purification of HCoV-19 spike ectodomain and hACE2》
2.1. Expression and purification of HCoV-19 spike ectodomain and hACE2
HCoV-19 S gene (GenBank number QHD43416.1) was synthesized (Genscript) with its codons optimized for insect cell expression. Its ectodomain (Val16-Pro1213) was cloned into a pFastBac vector (Life Technologies Inc., USA) with an N-terminal honeybee melittin signal peptide and C-terminal His6 and Flag tags. HCoV19 S protein was expressed in Sf9 insect cells using the Bac-toBac system (Life Technologies Inc.) and harvested from the cell culture medium; this was followed by a purification procedure using a Ni-nitrilotriacetic acid (NTA) column and Superdex 200 gel filtration column (GE Healthcare, UK) in tandem. The extracellular domain of hACE2 (Gln18-Ser740, NP_068576.1) with a Cterminal Fc tag was expressed in HEK 293 cells and was purified by protein A sepharose beads (GE Healthcare).
《2.2. Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDSPAGE) analysis》
2.2. Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDSPAGE) analysis
To test the glycosylation status of the HCoV-19 spike and the hACE2 protein, both proteins were deglycosylated by PNGase F (NEB, 1:50) overnight at 37 °C in phosphate-buffered saline (PBS). Both the glycosylated and deglycosylated forms of the HCoV-19 spike and the hACE2 protein were then analyzed by 15% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), followed by Coomassie blue staining.
《2.3. Glycopeptide sample preparation》
2.3. Glycopeptide sample preparation
The proteins were digested into peptides according to Ref. [31]. In brief, hACE2 and the S protein were first reduced by 10 mmol∙L–1 dithiothreitol in 50 mmol∙L–1 ammonium bicarbonate (ABC) for 45 min at room temperature and then alkylated by iodoacetamide for 45 min at room temperature in the dark. The proteins were then cleaned using acetone precipitation and resuspended in 50 mmol∙L–1 ABC. The alkylated glycoproteins were then digested for 14 h at 37 °C using sequencing-grade trypsin, chymotrypsin, or endoproteinase Lys-C (all purchased from Promega, USA), with a protein:protease ratio of 1:50 in 50 mmol∙L–1 ABC. The peptides were desalted using hydrophilic–lipophilic balance columns (Waters, USA).
The peptide samples were separated into duplicates. The first duplicate was deglycosylated by PNGase F (NEB, 1:100) overnight at 37 °C in 50 mmol∙L–1 ABC prepared in pure H2O18. The second duplicate was used to enrich intact N-glycopeptides by hydrophilic interaction liquid chromatography (HILIC). In brief, GlycoWorks (Waters) cartridges were preconditioned with loading buffer comprising 15 mmol∙L–1 ammonium acetate and 0.1% trifluoroacetic acid (TFA) in 80% acetonitrile. Peptides in the loading buffer were applied to the cartridges and the unbound flow-through fraction was collected. The column was washed two times with loading buffer. Glycopeptides were eluted in 0.1% TFA. All peptide fractions were desalted by house-made C18 stage tips before liquid chromatography–tandem mass spectrometry (LC-MS/MS) analyses were performed.
《2.4. Liquid chromatography–tandem mass spectrometry experiment》
2.4. Liquid chromatography–tandem mass spectrometry experiment
About 500 ng of peptide was analyzed using an Ultimate 3000 nanoflow liquid chromatography system (Thermo Scientific, USA) connected to a hybrid Q-Exactive HFX mass spectrometer (Thermo Scientific). The mass spectrometer was operated in data-dependent mode with full-scan MS spectrum, followed by MS2 scans recording the top 20 most intense precursors sequentially isolated for fragmentation using high-energy collision dissociation. The MS and MS/MS spectra were recorded using Xcalibur software 2.3 (Thermo Scientific). Detailed parameters for separation and MS acquisition can be found in Tables S1 and S2 in Appendix A, respectively.
《2.5. Bioinformatics》
2.5. Bioinformatics
The acquired MS raw files from the deglycosylated peptides were searched by MaxQuant (version 1.6.10.43; Max-PlanckInstitute of Biochemistry, Germany) against the hACE2 sequence from UniProtKB and the HCoV-19 S sequence (YP_009724390.1_3) from the National Center for Biotechnology Information (NCBI). Cysteine carbamidomethylation was set as a fixed modification, while methionine oxidation and O18 deamidation on glutamic acid were set as variable modifications. Trypsin was set as having up to two missed cleavages. A mass tolerance of 15 and 4.5 ppm was set for the first and main search, respectively. For comprehensive PTM analysis, phosphorylation (S/T/Y), acetylation (K), methylation (K/R/E), succinylation (K), crotonylation (K), farnesylation (K/N-terminal (Nterm)), myristoylation (K/Nterm), palmitoylation or prenylation, glycosylphosphatidylinositol addition, and oxidation on proline were individually investigated in parallel database searches for respective variable modification. A peptide-level false discovery rate of 1% was set to filter the result. Confident identification of PTM was based on a localization probability of 99%. Site occupancy for each PTM was calculated by dividing the peak intensities of the modified peptides (MP) and corresponding non-modified peptides (NP) using the following equation: MP/(MP + NP) ×100.
To investigate the N-glycosylation forms, the acquired MS raw files from the HILIC experiments were searched by pGlyco (version 2.2.2, Chinese Academy of Sciences, China) [32] against the same hACE2 and HCoV-19 S sequences. Cysteine carbamidomethylation was set as a fixed modification, while methionine oxidation was set as a variable modification. Trypsin was set as having up to two missed cleavages. A mass tolerance of 5 and 15 ppm was set for the precursor and fragment mass tolerance, respectively. Potential glycan fragments within the MS2 spectra were annotated by the built-in pGlyco.gdb glycan structure database [32]. The precursor intensity of each glycopeptide was extracted by the MaxQuant feature-detection algorithm. All LC-MS/MS raw data were deposited in an iProX database with the following link: https://www. iprox.cn/page/PSV023.html;?url=1631583654953TDY8, password: th2J; password: B73w.
《2.6. Structural model refinement》
2.6. Structural model refinement
Glycan structures in protein databank (PDB) format were downloaded or predicted by GLYCAM-Web (https://dev.glycam.org/gp). N-linked glycan models of hACE2 and the trimeric S protein were made by manually adding the MS-identified glycan at each site based on previous models (PDB codes 6M18 for hACE2 and 6VXX for S protein) within Coot [33]. The modified residues within each model were generated with Coot [33]. In addition, models of the HCoV-19 S protein and hACE2 complex (PDB 6M0J) were used to show the location of the PTMs surrounding the binding area. All structural analyses were performed by PyMOL (Schrödinger Inc., USA).
《2.7. Evaluation of binding of HCoV-19 S protein and hACE2 by biolayer interferometry》
2.7. Evaluation of binding of HCoV-19 S protein and hACE2 by biolayer interferometry
Binding of the HCoV-19 S protein and hACE2 was measured by bio-layer interferometry (BLI) on an Octet Re96E (ForteBio, USA) interferometry system. In brief, the HCoV-19 S protein was immobilized using aminopropylsilane biosensors (18-5045; ForteBio). To evaluate the influence of the glycosylation of HCoV-19 S protein on the binding, the S protein was first incubated with either 1000 U∙mL–1 PNGase F (NEB) or deactivated PNGase F at 37 °C for 14 h. After washing by kinetic buffer (1× PBS, pH 7.4, 0.01% bovine serum albumin (BSA), and 0.002% Tween 20), the association between HCoV-19 S protein and hACE2 was measured for 180 s at 30 °C by exposing sensors to hACE2 in kinetic buffer at concentrations of 6.25, 12.5, 25, 50, 100, and 200 nmol∙L–1 , respectively. After the binding phase, the sensors were exposed to 1 ×kinetic buffer for dissociation for 300 s at 30 °C. The signal baseline was subtracted before data fitting using the equimolar binding model. Mean association rate constant (kon) and dissociation rate constant (koff) were determined with a global fit model using all data. A parallel experiment was also performed using hACE2-loaded sensors incubated with HCoV-19 S protein solutions at different concentrations. Analogously, immobilized hACE2 was pretreated by either PNGase F or deactivated PNGase F to evaluate the influence of hACE2 glycosylation on binding. In all experiments, PNGase F was deactivated by heating at 75 °C for 10 min.
《3. Results》
3. Results
《3.1. Determination of N-linked glycosylation sites on HCoV-19 S protein and hACE2》
3.1. Determination of N-linked glycosylation sites on HCoV-19 S protein and hACE2
As shown in Fig. 1(a), PNGase F deglycosylation resulted in a decrease in the molecular weight of both HCoV-19 S protein and hACE2 on SDS-PAGE. Digestion by PNGase F releases N-linked glycans from Asn within the N-X-S/T motif, which also results in Asn deamidation by losing one hydrogen and one nitrogen while incorporating one oxygen derived from the solvent [34]. To perform a thorough survey of glycosylated sites, protease-digested peptides from both proteins were subjected to PNGase F deglycosylation in H2O18; this enables the incorporation of O18 and leads to a +2.98 Da mass increment, which was used to mark the glycosylated sites.
Analysis of the deglycosylated peptides from the S protein by LC-MS/MS confirmed the presence of 20 N-linked glycosylation sites, including N61, N74, N122, N165, N234, N282, N331, N343, N603, N616, N657, N709, N717, N801, N1074, N1098, N1134, N1158, N1173, and N1194 (Table 1). Two remaining N-X-S/T sites—the N17 and N149 sites—were not identified in any deglycosylated peptide. However, a peptide with glycosylated N149 was identified directly without PNGase F treatment, as described later, resulting in a total of 21 glycosylation sites out of 22 potential sites with N-X-S/T sequons in the HCoV-19 S protein (Fig. 1(b)). In addition, quantitative analysis of site occupancy showed that 18 sites were completely glycosylated, while N603 and N657 reached 43% and 74% occupancy, respectively (Table 1). Fig. 1(c) provides an example of mass spectra showing evidence of N331 and N343 glycosylation.
Quantitative analysis of site occupancy showed that all seven possible glycosylation sites in hACE2—namely, N53, N90, N103, N322, N432, N546, and N690—were completely glycosylated (Table 1). Fig. 1(d) provides an example of the mass spectra showing N90 glycosylation. These data suggest that both HCoV-19 S protein and hACE2 are highly decorated by N-glycans.
《Fig. 1》

Fig. 1. Potential glycosylation sites in the HCoV-19 S protein and hACE2. (a) 15% SDS-PAGE analysis of the intact and deglycosylated form of the HCoV-19 S protein and hACE2. Molecular weight markers are shown on the left. (b) Schematic representation of the functional subunits and domains of the HCoV-19 S protein (upper panel) and hACE2 (lower panel). CD: connector domain; CH: central helix; CT: cytoplasmic tail; FP: fusion peptide; TM: transmembrane domain; UH: upstream helix; HR1/2: heptad repeat 1/2. Blue indicates the domain or amino acids possibly responsible for interaction between the S protein and hACE2. Potential glycosylation sites within each domain are indicated. Red marks represent identified glycosylated sites in this study. (c, d) Mass spectra of deglycosylated peptide containing (c) N331 and N343 in the HCoV-19 S protein and (d) N90 in hACE2. NTD: N-terminal domain; PD: peptidase domain; CLD: C-terminal collectrin-like domain; de: deamidation.
《3.2. Identification of PTMs of HCoV-19 S protein and hACE2》
3.2. Identification of PTMs of HCoV-19 S protein and hACE2
To unveil possible PTMs other than glycosylation, LC-MS/MS data of deglycosylated peptides from both HCoV-19 S protein and hACE2 were subjected to several rounds of spectra-database matching for common PTMs. The major PTMs found in both proteins were methylation on lysine, arginine, or glutamic acid, as summarized in Table 1. Using an occupancy greater than 50% as a criterion to identify sites dominantly decorated with PTMs, we found that R78, E224, E654, and E661 in HCoV-19 S protein and E57, K68, and E329 in hACE2 were highly methylated. In addition, the proline at sites 253, 263, 321, and 346 in hACE2 was found to be prominently oxidized and converted to hydroxyproline. However, common PTMs such as phosphorylation, acetylation, or other forms of fatty acid acylations were not found in HCoV-19 S protein and hACE2.
《Table 1》
Table 1 Summary of PTM identified in deglycosylated peptides from HCoV-19 S protein and hACE2.



a Modified sites.
《3.3. Global N-glycosylation profile of HCoV-19 S protein and hACE2》
3.3. Global N-glycosylation profile of HCoV-19 S protein and hACE2
To resolve glycan camouflage on the surface of the HCoV-19 S protein and hACE2, intact glycopeptides derived from protease digestion and fractionated by HILIC were directly subjected to LC-MS/MS analysis specifically designed to detect peptides with extra molecular weight due to N-glycan attachment. Due to the highly heterogeneous nature of the glycan chain, a unique glycopeptide was designated in our study as a combination of both a unique peptide sequence and a specific N-glycan composition. According to these criteria, 419 and 467 unique N-glycopeptides were identified from the HCoV-19 S protein and hACE2, respectively (Tables 2 and 3). Except for N1134, 19 of the 20 HCoV-19 S glycosylated sides identified in the PNGase F experiment were confirmed by intact glycopeptide profiling. Furthermore, N-glycans were also found to present at site N149. Although all seven glycosylation sites in hACE2 were identified previously in the deglycosylated peptides labeled with deamidation, an N-glycan profile was obtained at five sites: N90, N103, N432, N546, and N690. Fig. S1 in the Appendix A provides an example of N-glycopeptides spectra from HCoV-19 S protein and hACE2.
A total of 144 N-glycans were found in the HCoV-19 S protein, with the majority containing the common N-acetylglucosamine core (Table 2). All N-glycosites in the S protein were attached to multiple types of N-glycans, with N343 being decorated by the most diverse N-glycans. The HCoV-19 S N-glycan composition was preferentially comprised of pauci- or high-mannose oligosaccharides, except in four N-glycosites (N73, N343, N717, and N1173) that contained a high proportion of complex and hybrid N-glycans (Figs. 2(a) and (b) and Table 2). Based on LC-MS intensity, pauci-mannose Hex3HexNAc2 was the most common Nglycan in the HCoV-19 S protein. Interestingly, there was no evidence of a sialic acid component in the N-glycans of the HCoV-19 S protein.
《Table 2》
Table 2 Summary of glycopeptide and N-glycan identification in HCoV-19 S protein.



a Glycosylated N sites.
b Sites identified only on deglycosylated peptides.
A total of 220 N-glycans were found in hACE2; of these, 93% were complex, while hybrid, high-mannose, and pauci-mannose glycan only constituted 4%, 2%, and 1%, respectively (Fig. 2(a) and Table 3). Based on LC-MS intensity, the most dominant hACE2 glycan was NeuGc1Hex5HexNAc4, which was predicted to be a biantennary complex containing a sialic acid in the distal end. The N-glycan profile in all hACE2 sites was primarily complex. It is notable that N90, N103, and N690 all contained more than 100 types of N-glycans, with their most dominant forms all containing sialic acid.
《Table 3》
Table 3 Summary of glycopeptide and N-glycan identification in hACE2 protein.

a Sites identified only on deglycosylated peptides.
b Glycosylated N sites.
《Fig. 2》
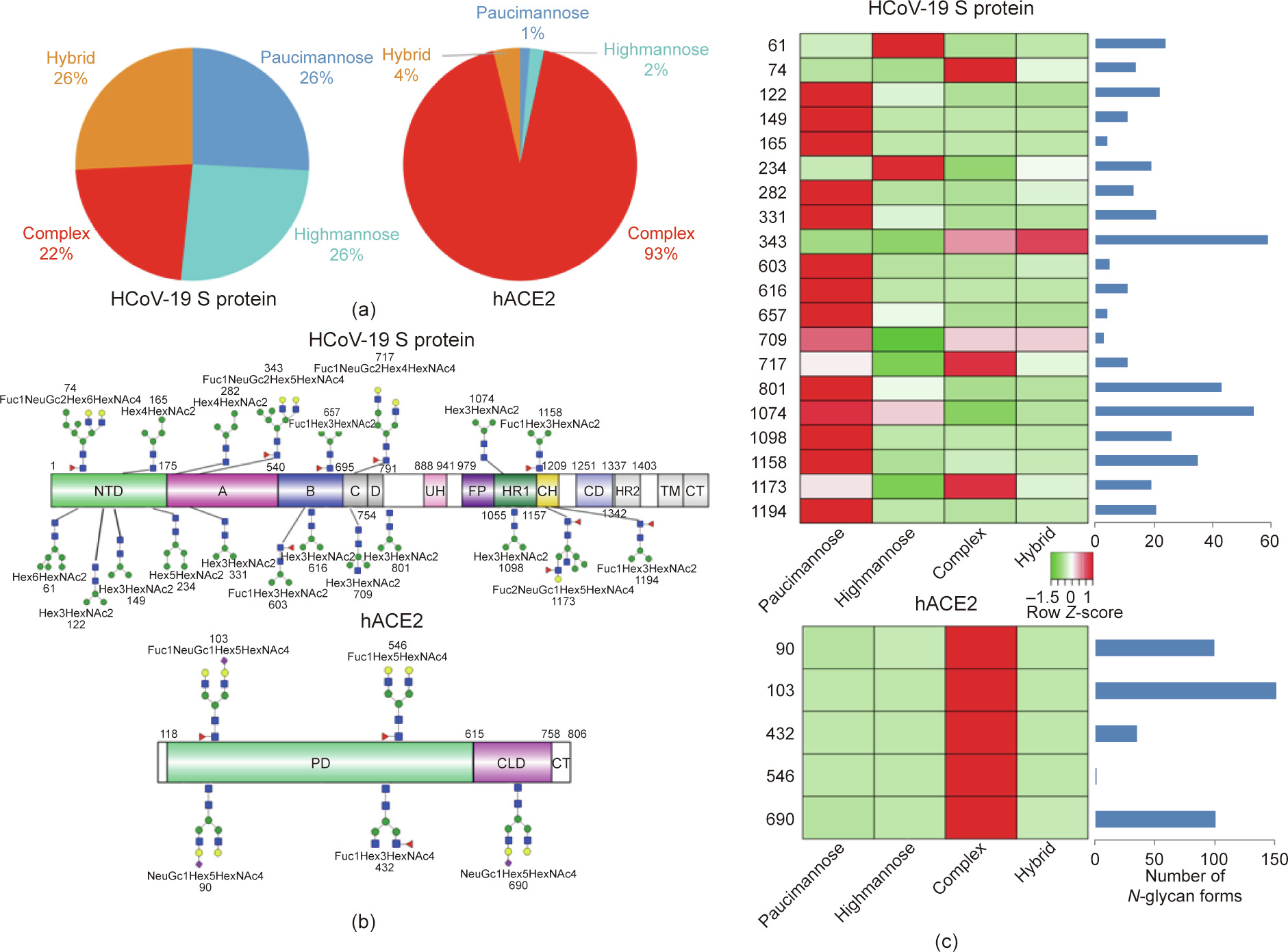
Fig. 2. Summary of an N-glycopeptide survey in the HCoV-19 S protein and hACE2. (a) Overall percentages of four major N-glycan categories identified in either protein. (b) Schematic representation of the most predominant N-glycan form in each site related to functional domains in either protein. (c) Relative LC-MS intensities of the four major N-glycan categories identified in each site presented as a heatmap, along with the total number of identified glycans, summarized in bar plots.
Collectively, our LC-MS/MS data confirm that both the HCoV-19 S protein and its receptor hACE2 are indeed heavily N-glycosylated at most of their predicted N-X-S/T sequons. Fig. 2(c) provides a summary of the most dominant N-glycan composition and predicted structure for each site of the HCoV-19 S protein and hACE2.
《3.4. Structure modeling of the HCoV-19 S–hACE2 complex refined with glycan and PTM details》
3.4. Structure modeling of the HCoV-19 S–hACE2 complex refined with glycan and PTM details
By adding the chemical structures of the most abundant N-glycans at each site, based on the LC-MS/MS results, to the most updated cryo-electron microscopy (cyro-EM) model of the HCoV19 S protein and hACE2, we generated atomic models that represent the most likely spatial distribution of the N-glycans on both proteins (Figs. 3(a) and (b)). Although four sites (N74, N149, N1158, and N1194) of the S protein are not shown in the model due to the missing residues in model 6VXX, our model suggests that the camouflaging N-glycans shield more than two-thirds of the HCoV-19 S protein surface, which could potentially lead to host attachment and immune evasion. In addition, both the HCoV-19 S protein and the hACE2 model showed that the glycans at N331 and N343 of the HCoV-19 S protein and N90 of hACE2 were in proximity to—albeit not exactly inside of—the binding area of both proteins (Figs. 3(c) and (d)). The model of the S protein–hACE2 complex also showed that three methylation sites in hACE2 (E57, K68, and E329) form a trident structure enclosing the contact area formed between K353–R357 of hACE2 and N501 of HCoV-19 S protein (Fig. S2 in the Appendix A).
《Fig. 3》
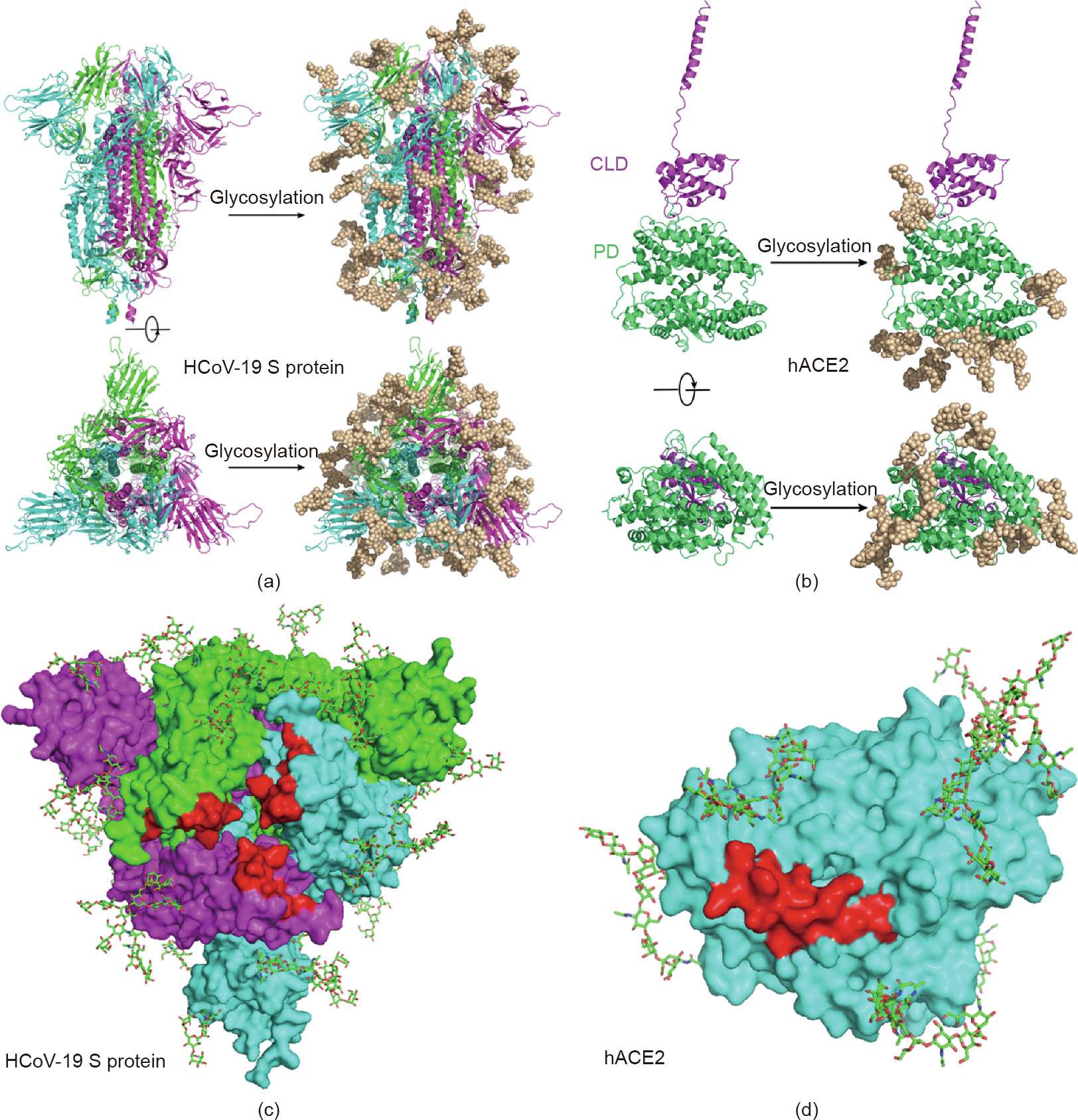
Fig. 3. Refined structural model of the HCoV-19 spike trimer and hACE2 incorporating N-glycans. (a, b) 3D ribbon diagrams of (a) the HCoV-19 spike trimer colored by protomer and (b) hACE2 colored by major domains. Side view (upper panel) and top view looking toward the viral or cellular membrane (lower panel) of both models without (left panel) or with glycans (right panel) are provided. (c, d) Diagrams showing binding sites (red) with glycans in the vicinity on the surface of (c) the HCoV-19 spike trimer and (d) hACE2 from the top view. Glycans are presented as sticks.
《3.5. Binding of HCoV-19 S protein and hACE2 does not depend on N-glycosylation》
3.5. Binding of HCoV-19 S protein and hACE2 does not depend on N-glycosylation
To understand the contribution of protein glycosylation to the interaction between HCoV-19 S protein and hACE2, we compared the binding kinetics and affinity of the purified hACE2 ectodomain to glycosylated and deglycosylated HCoV-19 S extracellular domain (ECD) immobilized at the surface of biosensors in a BLI experiment. We found that hACE2 was bound to HCoV-19 S protein that had been pretreated with active or inactive PNGase F with the comparable equilibrium dissociation constants Kd of 1.7 nmol∙L-1 (Fig. 4(a)) and 1.5 nmol∙L-1 (Fig. 4(b)), respectively. The affinity of hACE2 binding to HCoV-19 S protein that was determined in this study aligns with the previous report [13]. Comparable affinity was also observed when HCoV-19 S ECD was bound to the immobilized hACE2 (Kd = 16.7 nmol∙L-1 , Fig. 4(c)) or deglycosylated hACE2 (Kd = 18.2 nmol∙L-1 , Fig. 4(d)). Therefore, our data suggest that the glycosylation status does not contribute directly to the binding between HCoV-19 S protein and hACE2. A detailed summary of the BLI binding kinetics can be found in Table S3 in Appendix A.
《Fig. 4》
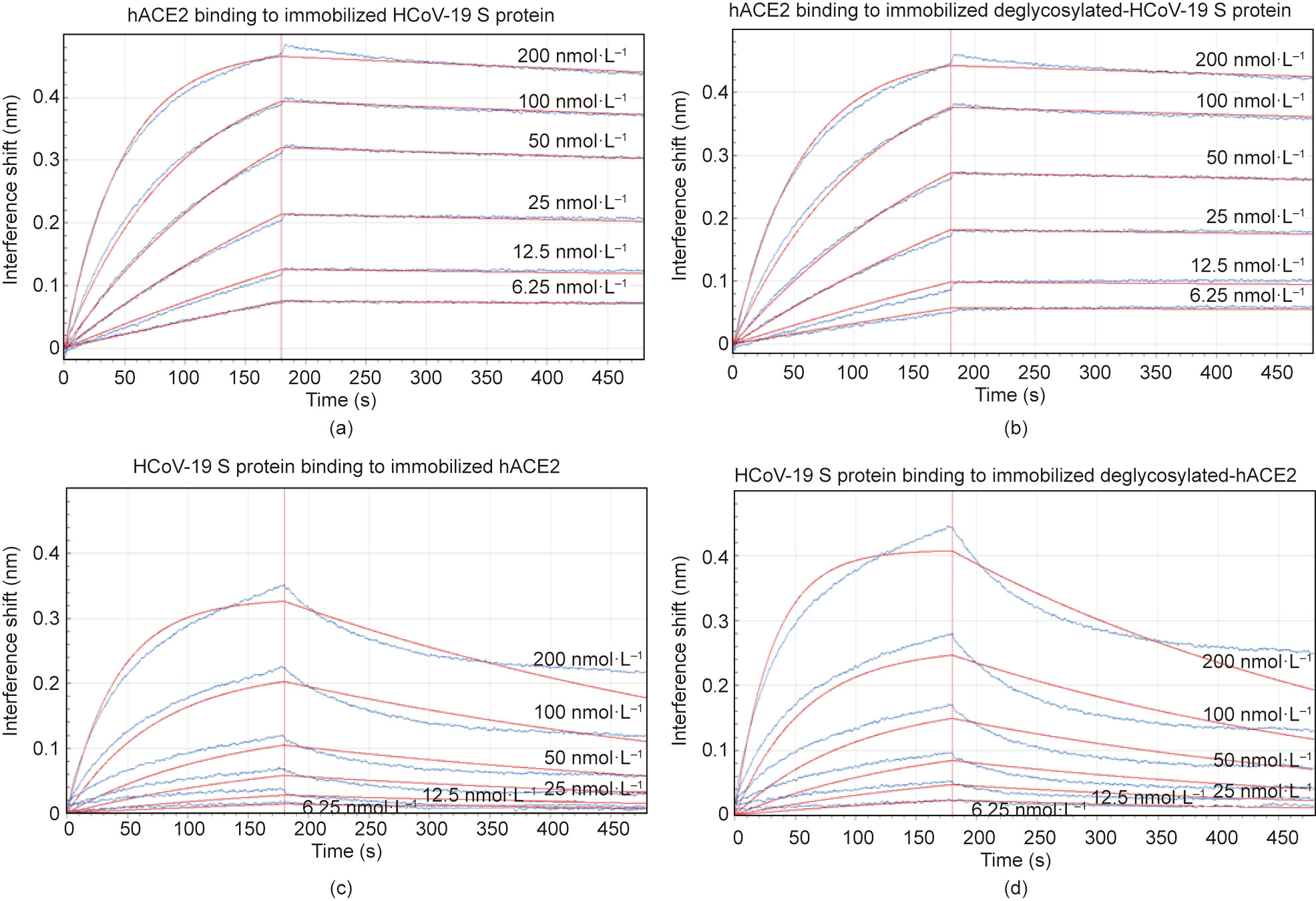
Fig. 4. Impact of glycosylation on binding between the HCoV-19 S protein and hACE2. Binding of the HCoV-19 S protein and hACE2 was measured by bio-layer interferometry. Biosensors with (a) immobilized intact HCoV-19 S protein and (b) its deglycosylated form were exposed to hACE2 at six concentration levels. Swap experiments were conducted using biosensors with (c) immobilized intact hACE2 and (d) its deglycosylated form exposed to HCoV-19 S protein at six concentration levels. Red lines correspond to a global fit of the data using an equimolar binding model.
《4. Discussion》
4. Discussion
Glycosylation is an ubiquitous and complex PTM that greatly extends the structural and functional diversity of many proteins. However, comprehensive characterization of protein N-glycosylation is technically challenging. In this study, deglycosylated peptides, which are easier to be analyzed by LC-MS than their glycosylated precursors, were profiled to confirm the N-glycosylated sites in HCoV-19 S protein and hACE2. The O18 deamidation modification ensured confidence in the glycosite identifications, as the isotope was incorporated during PNGase Fmediated hydrolysis in H2O18. Intact N-glycopeptide analyses were then performed to further verify the glycosites and to provide detailed information about the compositional and structural heterogeneity of the associated N-glycans in each site. Our investigation showed that all sites except N17 were highly glycosylated in the HCoV-19 S protein. According to the N-linked glycan model of the S protein, a large proportion of the S protein surface is covered by glycans, which is consistent with previous reports [13,14,35,36]. When the S proteins from HCoV-19 and SARS-CoV were compared, it was noticeable that the majority of differences in the glycosylation sites occurred in the distal S1 subunit, resulting in a significant difference in the glycan profile in the outermost canopy of the virus formed by spike trimer clusters. This alteration in glycosites might be the result of host or environmental selective pressure, and could imply dramatic differences in viral infectivity, pathogenesis, and host responses. During our research, we noticed that several other research groups also made attempts to decode the glycan profile of the HCoV-19 S protein. All these studies have confirmed that the HCoV-19 S protein is heavily glycosylated [35,36].
The N-glycans in the S protein are markedly heterogeneous and may greatly extend the conformational flexibility and epitope diversity for the S protein. Although a small proportion of complex and hybrid N-glycans were found in the S protein, most sites were occupied by oligomannose glycans, which is in line with previous reports on the glycosylation profile of SARS-CoV [37–39]. Many viral proteins are hallmarked by a high level of oligomannose glycans, probably as a sign of incomplete glycan maturation due to a high glycosite density that results in steric hindrance [40]. Interestingly, N343, the glycosite closet to the binding surface, is decorated by the most diverse N-glycans and primarily by hybrid and complex forms. Moreover, as in the case of SARS-CoV and Marburg viral proteins [41,42], we found that sialic acid incorporation in the SARS-CoV S protein glycans was negligible. In the ongoing global efforts to generate a vaccine, which primarily targets the S protein as the candidate antigen, vaccine developers should consider the diverse glycan forms decorating a large swath of the S trimer surface, as glycans could drastically modulate the protein immunogenicity. It was interesting to learn that different expression systems could affect glycan forms substantially, as we compared our data with other glycan profiling studies [35,36]. In particular, mammalian cells prefer to assemble complex glycans to the S protein, as compared with insect cells. Therefore, we should also expect that vaccines that are produced based on different S proteoforms with different glycosylation statuses could show varying efficacy against future infection.
The binding of coronavirus S proteins to their respective receptors has been shown to be mediated by viral oligomannose N-glycan [19,20,23]. The SARS-CoV S protein contains three RBDassociated glycosites (N318, N330, and N357), while HCoV-19 only has two (N331 and N343) surrounding the binding pocket. Contrary to our postulation that the N-glycan at these two sites might contribute polar interaction to receptor binding, the BLI binding assay demonstrated that deglycosylation did not change the affinity of the SARS-CoV S protein to hACE2. However, this negative BLI binding result does not exclude the possibility that glycosylation could affect other viral entry steps, including protease cleavage and glycan-mediated interactions with dendritic cell/ liver/lymph node-specific ICAM-3-grabbing non-integrin, which were reported to enhance SARS-CoV entry [25,26]. Further investigations are required to ascertain the role of glycosylation in the infectivity of HCoV-19.
Antigen glycosylation greatly determines host immune responses. One of the prominent consequences of glycosylation is immune evasion by shielding off immunogenic epitopes in viral envelope or surface proteins [17,43,44]. The complete occupancy of glycans in most of the glycosites of the HCoV-19 S protein suggests that the virus is able to invade the host in a stealthy fashion. Successful innate immune evasion by HCoV-19 during the early stage of infection might explain the long asymptomatic incubation period during which transmissible virions are produced [4,6]. Cases of HCoV-19 reinfection have also been reported, suggesting that the virus is capable of escaping from antibody-mediated neutralization [45]. On the other hand, protein glycosylation can also facilitate immune response by means of sensor molecules or anti bodies that are specific for glycan recognition. N-glycans are known ligands for galectins, and can trigger inflammation events such as cytokine release and immune cell infiltration [46,47]— which may contribute to the HCoV-19 pathogenesis and correlate with the disease severity. Moreover, epidemiological studies have shown that ABO polymorphism is linked to different susceptibilities to both HCoV-19 and SARS-CoV infections [48,49]. Since the ABH carbohydrate epitopes and viral protein glycans are likely to be synthesized by the same ER-resident glycosylation enzymes, anti-A or -B antibodies can partially block the viral–host interactions [50]; thus, individuals with blood type O would be more resistant to virus infection. It remains to be explored whether the glycosylation status of the S protein has implications toward inter-individual variation in terms of viral response and clinical outcome within the infected population.
The glycosylation status of hACE2 was also profiled in this study. All seven glycosites in hACE2 were completely occupied by glycan, including N90, which is in the vicinity of the binding surface. However, we found that glycans did not directly contribute to the binding of hACE2 with the HCoV-19 S protein. This finding is in agreement with a previous finding that disruption of ACE2 glycosylation did not affect its binding with the SARS-CoV S protein [51]. However, deglycosylated ACE2 did compromise the cellular entry and the subsequent production of infectious SARSCoV virion [51]. Therefore, ACE2 glycosylation is still considered to be an important target for intervention of coronavirus infection. In fact, the ability of chloroquine to counter HCoV-19 infection is thought to be due to its inhibition of ACE2 glycosylation, in addition to its ability to increase the endosomal pH level [28,52].
In addition to glycosylation, additional PTM forms were investigated in this study. Methylation was identified in several sites in both the S protein and hACE2. In particular, we found that the E57, K68, and E329 sites in hACE2, which surround its binding site with the RBD of the S protein, are completed methylated. Methylation causes a loss of charge and increases the hydrophobicity of these sites. Four hydroxyprolines (at 253, 263, 321, and 346) were identified in the protease domain of hACE2. The extra hydroxyl group is likely to increase the hydrophilicity of proline in this extracellular region of hACE2. Further study is needed to investigate possible biological roles of these PTMs. We did not find phosphorylation or acetylation in our dataset, and previous PTM investigation of SARS-CoV also failed to identify these PTMs [38]. Fatty acid acylations were also considered in our study, since they are commonly found in surface viral proteins to facilitate membrane fusion and virus entry. However, our study found no evidence of an acylation PTM presence in the HCoV-19 S protein. Given that our multi-protease proteomic experiment provides almost full coverage for both proteins, we believe that the PTMome of the HCoV-19 S protein and hACE2 mainly consists of glycosylation, methylation, and proline oxidation.
《Acknowledgments》
Acknowledgments
This work was supported by the National Key Research and Development Program (2017YFC1200204, 2017YFA0504803, and 2018YFA0507700), Emergency Project of Zhejiang Provincial Department of Science and Technology (2020C03123-1), Fundamental Research Funds for the Central Universities (2018XZZX001-13), and Independent Project Fund of the State Key Laboratory for Diagnosis and Treatment of Infectious Disease. We thank the proteomics and metabolomics platform in the State Key Laboratory for Diagnosis and Treatment of Infectious Diseases at Zhejiang University for glycoproteomic analysis. We thank Shuangtian Shengwu Biotech. Co., Ltd. for assistance of BIL analysis. We thank pGlyco team at Institute of Computing Technology (Chinese Academy of Sciences, Beijing, China) for technical support for pGlyco analysis. We thank Dr. Ma Ping for consultation on protein expression and purification.
《Authors’ contributions》
Authors’ contributions
Study concept and design: Zeyu Sun, Lanjuan Li. Samples preparation: Zeyu Sun, Keyi Ren, Feiyang Ji, Xiaoxi Ouyang. LC-MS/MS experiments: Zeyu Sun, Keyi Ren. Analysis and interpretation of data: Zeyu Sun, Jing Jiang, Keyi Ren. Structure modeling: Xing Zhang, Jinghua Chen, Zeyu Sun. Drafting of the manuscript: Zeyu Sun, Zhengyi Jiang. Critical revision of the manuscript: Lanjuan Li. All authors approved the final version of the manuscript, including the authorship list.
《Compliance with ethics guidelines》
Compliance with ethics guidelines
Zeyu Sun, Keyi Ren, Xing Zhang, Jinghua Chen, Zhengyi Jiang, Jing Jiang, Feiyang Ji, Xiaoxi Ouyang, and Lanjuan Li declare that they have no conflict of interest or financial conflicts to disclose.
《Appendix A. Supplementary data》
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.eng.2020.07.014.











 京公网安备 11010502051620号
京公网安备 11010502051620号




