

《1. Introduction》
1. Introduction
With an indispensable role in modern therapeutics for diseases such as infection, inflammatory diseases, and cancers, therapeutic antibodies contribute to the incomparably rapid research and development (R&D) of biopharmaceuticals. As of 31 December 2020, 106 therapeutic antibodies have been approved by the US Food and Drug Administration (FDA). Furthermore, in the past two decades, global sales of antibody products have increased around 450-fold—from 0.31 billion USD in 1997, to 37 billion USD in 2008, and then to approximately 135 billion USD in 2018. Despite the prevalent application of therapeutic antibodies, drawbacks have been occurring, including unsatisfactory efficacy, which is attributed to low affinity of antibodies; weak potency of inducing/induced effector functions; unexpected degradation in circulation or intracellular catabolism; severe systemic toxicity caused by inevitable binding to non-target cells expressing targeted antigens; and exposure to large dosages of therapeutics. Therefore, antibody engineering is indispensable in order to modify developing and developed therapeutic antibodies through approaches such as glycoengineering technology and mutagenesis, thereby optimizing therapeutic efficacy to delay the progression of diseases.
As the molecular mechanisms of disease onset and subtle regulatory networks are unveiled, scholars have embarked on modifications of therapeutic antibodies for precise and industrialized treatment and magnified effectiveness. Modified therapeutic antibodies refer to antibodies based on the specific binding of variable regions; they show overall improved efficacy to indications, which is reflected by reduced mortality (in protective therapy) or morbidity (in preventative therapy), impeded relapse, protection from adverse reactions, and so forth. At the molecular level, even though the engagement of antibodies and antigens is fundamental to the efficacy of therapeutic antibodies, interactions between constant regions and corresponding receptors on host cells are nonnegligible, since they directly modulate the effector functions of immune responses by recruiting and activating effector cells and through the intracellular degradation and recycling of therapeutic antibodies. Each biological process can be regarded as a target for the modification of therapeutic antibodies to promote efficacy. This review mainly focuses on the progress that has been achieved in six common approaches to therapeutic antibody modification. The accomplishments of China in this realm are also summarized.
《2. An overview of the evolution of antibody theory and structure-based functions》
2. An overview of the evolution of antibody theory and structure-based functions
In 1888, antibodies were applied to therapy for the first time by Emile Roux [1], who administered polyclonal antisera to cure diphtheria. In 1897, Ehrlich hypothesized that antibody binding with specific chemicals, metaphorized as a ‘‘magic bullet,” could be utilized for targeted therapy, considering the specificity of antibodies [2]. Von Behring and Kitasato then put forward humoral immunity theory based on antibodies in the early 19th century [3]. The structure of the antibody was discovered by Porter in 1959 [4]; Tonegawa et al. [5] then illustrated the genetic basis of antibody variability in 1974. Meanwhile, the revolutionary technology of hybridoma cells was invented by Köhler and Milstein, providing access to the design and manufacturing of therapeutic antibodies [6]. These milestone achievements laid the foundation of antibody engineering for therapy. Since the first therapeutic antibody, orthoclone OKT3, was approved by the FDA in 1986, an increasing number of antibody drugs have entered the biopharmaceutical market and have significantly motivated the biopharma industry. R&D toward modified therapeutic antibodies has been emerging in recent years, evoked by the limitations of current treatment and the assistance of biomedical techniques such as next-generation DNA sequencing (NGS), proteomics, gene editing, and computeraided screening. In the foreseeable future, modified therapeutic antibodies will play a greater role in therapies and occupy a larger portion of the biopharma industry (Table 1).
《Table 1 》
Table 1 List of modified therapeutic antibodies and recent-five-year approved therapeutic antibodies on market by FDA.



CD: cell differentiation molecular; Fc: fragment of crystallizable domain; ADC: antibody–drug conjugate; CCR: CC chemokine receptor; ALL: acute lymphatic leukemia; HER: human epidermal growth factor; VEGFR: vascular endothelial growth factor receptor; PD-1: programmed cell death receptor-1; IL: interleukin; GD: disialoganglioside; PCSK: proprotein convertase subtilisin/kexin; EGFR: epidermal growth factor receptor; SLAMF: signaling lymphocytic activation molecule family; PD-L1: programmed cell death-ligand 1; PDGFRα: platelet-derived growth factor receptor alpha; HIV: human immunodeficiency virus; FGF: fibroblast growth factor; CGRP: calcitonin gene-related peptide; VEGF: vascular endothelial growth factor; aTTP: acquired thrombotic thrombocytopenic purpura; CLL: chronic myeloid leukemia; CSCC: cutaneous squamous cell carcinoma; HCL: hairy cell leukemia; MCC: Merkel cell carcinoma; MM: multiple myeloma; MS: multiple sclerosis; NSCLC: non-small cell lung carcinoma; PNH: proxysmal nocturnal hemoglobinuria; RA: rheumatoid arthritis; r/r B-ALL: relapsed or refractory B cell precursor acute lymphoblastic leukemia; r/r DLBL: relapsed or refractory diffuse large B cell lymphoma; XLH: X-linked hypophosphatemia; vWF: von Willebrand factor.
As important effectors of immune response, antibodies can recognize, neutralize, and clear pathogenic antigens. These functions are regulated by structure-determined properties such as binding affinity and pharmacokinetics. In structural and functional research, an antibody can be hydrolyzed by papain under certain conditions to yield two fragments of antigen binding (Fab) and one fragment of crystallizable domain (Fc). More specifically, a Fab consists of one variable region of heavy chain (VH), one variable region of light chain (VL), one constant region of light chain (CL), and one heavy chain constant region 1 (CH1). In VH and VL, respectively, there are three different variable amino acid sequences that contribute to the spatial conformation complementary to the epitopes of antigens. These sequences are called the complementary determining regions (CDRs), and the rest is called the framework region (FR). The Fab domain specifically discriminates and binds antigens or targeted cells in a monovalent pattern. As Fab modification, the site-directed mutagenesis of histidine (with –lgKa around 6,Ka is the dissociation constant of the acid) residues into CDRs or FR has yielded pH-dependent binding antibodies that dissociate the bound antigen in acidic endosomes and can be recycled back to plasma to bind antigens more than once [7–9]. This modification strategy has been applied in satralizumab against interleukin-6 receptor (IL-6R) for neuromyelitis optica spectrum disorder (NMOSD), and demonstrated an association with a lower risk of relapse [10]. The Fc portion, which consists of CH2 and CH3 domains without the activity of binding antigens, interacts with the receptors (FcRs) on target cells and subsequently initiates multiple immunological effects, such as antibody-dependent cellular cytotoxicity (ADCC). Fc modification, such as engineered afucosylation, enhances ADCC and consequently improves the therapeutic potency of antibodies such as the marketed mogamulizumab against C–C chemokine receptor type 4 (CCR4) for T cell leukemia. In addition, the antibody receptor is crucial for binding and for the downstream biological process. The neonatal Fc receptor (FcRn) on the target cells regulates immunoglobulin G (IgG) catabolism which relies on pH [11,12]. At low pH (pH < 6.5), binding is regulated by the protonation of histidine 310 (His310), His435, and His436 in the Fc region [13]. Protonation results in residues with a positive charge that can bind to glutamic acid 117 (Glu117), Glu132, and aspartic acid 137 (Asp137) with a negative charge in the FcRn [14]. In the physiological extracellular environment, the weak affinity of IgG to FcRn leads to its release from the receptor into circulation [15]. These structural characteristics can not only be naturally regulated, but also be artificially modified, in order to ensure the clinical effectiveness of therapeutic antibodies as biopharmaceuticals.
《3. Progress in modified therapeutic antibodies around the world》
3. Progress in modified therapeutic antibodies around the world
As the diversity of innovative antibody drugs is continually increasing around the world, modifications of therapeutic antibodies have become a hot topic in biomedical research. In terms of function, antibodies recruit immune components from patients to carry out effector functions, the major mechanisms of which include ADCC, complement-dependent cytotoxicity (CDC), and antibody-dependent cellular phagocytosis (ADCP) [16]. The involvement of Fc gamma receptors  is necessary for ADCC and ADCP, as the complement protein 1q (C1q) of the complement system bound to the antibody CH2 domain activates CDC. Moreover, IgG is endocytosed by cells, where it can be shuttled from endosomes to lysosomes for degradation with low FcRn binding affinity [15] or recycled back to the extracellular membrane [17,18]. Determined by the antibody–FcRn binding, the circulation and half-life can be improved by modification to ameliorate the disadvantages in pharmacodynamics. Based on researchers’ efforts to modify therapeutic antibodies, modifications can be categorized as follows: ① modified glycosylation; ② Fc amino acid alterations; ③ cross-isotype or cross-subclass exchanges; ④ antibody–drug conjugates (ADCs); ⑤ single chain of variable region fragment (scFv) for chimeric antigen receptor T (CAR-T) cells; and ⑥ bispecific antibodies (bsAbs). Progress that has been achieved in modified therapeutic antibodies is summarized below, according to this categorization (Fig. 1).
is necessary for ADCC and ADCP, as the complement protein 1q (C1q) of the complement system bound to the antibody CH2 domain activates CDC. Moreover, IgG is endocytosed by cells, where it can be shuttled from endosomes to lysosomes for degradation with low FcRn binding affinity [15] or recycled back to the extracellular membrane [17,18]. Determined by the antibody–FcRn binding, the circulation and half-life can be improved by modification to ameliorate the disadvantages in pharmacodynamics. Based on researchers’ efforts to modify therapeutic antibodies, modifications can be categorized as follows: ① modified glycosylation; ② Fc amino acid alterations; ③ cross-isotype or cross-subclass exchanges; ④ antibody–drug conjugates (ADCs); ⑤ single chain of variable region fragment (scFv) for chimeric antigen receptor T (CAR-T) cells; and ⑥ bispecific antibodies (bsAbs). Progress that has been achieved in modified therapeutic antibodies is summarized below, according to this categorization (Fig. 1).
《Fig 1》

Fig 1. Common modification approaches to modify therapeutic antibodies. (a) Modified glycosylation targets the fucosylation site asparagine 297 (Asn297) in the CH2 domain of the Fc region. The key enzymes and transporters are major regulators of fucosylation, including 4,6-dehydratase (GMD), guanosine diphosphate (GDP)-keto-6- deoxymannose 3,5-epimerase/4-reductase (FX), α-1,6-fucosyltransferase (FUT8), and GDP-fucose transporter (GFT). Using a loss-of-function strategy toward them is a routine afucosylation modification. The dashed line represents variable glycosylation, while the solid line represents core glycosylation in the Asn297 glycans of the Fc region. (b) Fc amino acid alterations can change the affinity and effector functions of target cells and the half-life of antibodies. The serine 239 aspartic acid (Ser239Asp)/isoleucine 332 glutamic acid (Ile332Glu)/alanine 330 leucine (Ala330Leu) termed DLE mutation is shown as an example. (c) A cross-isotype antibody can integrate different effector functions elucidated by different isotypes of immunoglobulins. In the fusion regions in the Fc domain of the illustrated example, the cross-isotype IgGA consists of CH1a– CH2g–CH3g–CH3a. (d) An ADC is a specific antibody-guidance-based cytotoxic drug. Here, the ten-angled star represents radioisotope 131I, the pentagon in light blue represents the dextran polysaccharide scaffold, and the oval in dark blue represents the cytotoxic agent. (e) The CAR illustrated here is a third-generation CAR with both costimulatory domains CD28 and anti-human-tumor necrosis factor receptor superfamily member 9 (4-1BB) in the intracellular membrane. The VH and VL depend on specific antibodies targeting the corresponding antigens. (f) bsAbs vary considerably in terms of component and format, and have integrated functions. NK: natural killer.
《3.1. Modified glycosylation》
3.1. Modified glycosylation
ADCC is triggered by the interactions of Fc with the corresponding 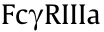 (CD16a) expressed on natural killer (NK) cells, which is sensitive to the state of Fc glycosylation [19,20]. The Fc region of IgG includes an N-linked glycosylation site at position asparagine 297 (Asn297), where the oligosaccharide chain is usually composed of two N-acetylglucosamine (GlcNAc), three mannose in a ‘‘V” pattern, and two GlcNAc linked to the mannose to form a biantennary complex glycan [21]. Additional fucose, GlcNAc, sialic acid, and galactose can be connected to the core glycan structure. With direct effects on
(CD16a) expressed on natural killer (NK) cells, which is sensitive to the state of Fc glycosylation [19,20]. The Fc region of IgG includes an N-linked glycosylation site at position asparagine 297 (Asn297), where the oligosaccharide chain is usually composed of two N-acetylglucosamine (GlcNAc), three mannose in a ‘‘V” pattern, and two GlcNAc linked to the mannose to form a biantennary complex glycan [21]. Additional fucose, GlcNAc, sialic acid, and galactose can be connected to the core glycan structure. With direct effects on  binding, the Asn297 glycan (which is actually fucose at this site) can clash with glycans on CD16a and influence hinge region conformations, which attenuates the engagement of the effector cells in ADCC [22–24]. Although the majority of mammalian IgG is fucosylated at Asn297 [25–27], strategies have been put forward based on a comprehensive understanding of this site-specific fucosylation in the Fc region. The fucosylation process contains three steps: ① Guanosine 5' -diphospho-β-L-fucose (GDP-fucose) synthesized in the cytoplasm is the substrate of the first step, and is derived from GDP-mannose. The conversion to GDP-fucose is catalyzed by 4,6-dehydratase (GMD) and GDP-keto-6-deoxymannose 3,5-epimerase/4-reductase (known as FX) as the major anabolic source, known as the de novo pathway [28]. ② As the substrate of fucosylation reactions, the synthesized GDP-fucose must be transported to the endoplasmic reticulum (ER) or Golgi apparatus. The GDP-fucose transporter (GFT) encoded by the Slc35c1 gene is responsible for transportation to the Golgi apparatus [29]. ③ FUT8 is the sole α-1,6-fucosyltransferase to transfer fucose via an α-1,6 linkage to the innermost GlcNAc on N-glycans for core fucosylation [30]. Furthermore, the generation of bisecting GlcNAc catalyzed by β-1,4-mannosyl-glycoprotein 4-β-N-acetylglucosaminyltransferase (GnT-III) is effective to induce afucosylation because a GlcNAc is attached through a 1,4-β-linkage to the β-linked mannose of the N-glycan trimannosylated core [31]. Thus, targeting the abovementioned enzymes to regulate the fucosylation process is theoretically feasible for afucosylation to enhance the ADCC effect.
binding, the Asn297 glycan (which is actually fucose at this site) can clash with glycans on CD16a and influence hinge region conformations, which attenuates the engagement of the effector cells in ADCC [22–24]. Although the majority of mammalian IgG is fucosylated at Asn297 [25–27], strategies have been put forward based on a comprehensive understanding of this site-specific fucosylation in the Fc region. The fucosylation process contains three steps: ① Guanosine 5' -diphospho-β-L-fucose (GDP-fucose) synthesized in the cytoplasm is the substrate of the first step, and is derived from GDP-mannose. The conversion to GDP-fucose is catalyzed by 4,6-dehydratase (GMD) and GDP-keto-6-deoxymannose 3,5-epimerase/4-reductase (known as FX) as the major anabolic source, known as the de novo pathway [28]. ② As the substrate of fucosylation reactions, the synthesized GDP-fucose must be transported to the endoplasmic reticulum (ER) or Golgi apparatus. The GDP-fucose transporter (GFT) encoded by the Slc35c1 gene is responsible for transportation to the Golgi apparatus [29]. ③ FUT8 is the sole α-1,6-fucosyltransferase to transfer fucose via an α-1,6 linkage to the innermost GlcNAc on N-glycans for core fucosylation [30]. Furthermore, the generation of bisecting GlcNAc catalyzed by β-1,4-mannosyl-glycoprotein 4-β-N-acetylglucosaminyltransferase (GnT-III) is effective to induce afucosylation because a GlcNAc is attached through a 1,4-β-linkage to the β-linked mannose of the N-glycan trimannosylated core [31]. Thus, targeting the abovementioned enzymes to regulate the fucosylation process is theoretically feasible for afucosylation to enhance the ADCC effect.
To modulate the enzymes related to fucosylation, modification of the production cell lines serves as a common approach by means of gene editing such as knock down/out or zinc-finger nucleases (ZFNs). Louie et al. [32] reported an FX-knockout Chinese hamster ovary (CHO) cell line that could be used to produce antibodies with completely afucosylated N-glycans. Inactivation of the Slc35c1 and FUT8 gene by ZFNs in an industrial protein-production host cell line CHO yielded afucosylated antibodies without detrimental effects on cell growth, viability, or product quality [31]. Two of the three marketed glycoengineering antibodies—namely, mogamulizumab (Poteligeo) and benralizumab (Fasenra, MEDI-563)— are produced in FUT8-knockout CHO cells. Furthermore, the combination of GnT-III and Golgi resident enzyme α-mannosidase II (αManII) overexpression was found to bring about the highest level of bisecting and afucosylated glycans on IgG antibodies, and has been translated into the commercialized anti-CD20 antibody obinutuzumab (GA101). Obinutuzumab has enhanced affinity and is the first line of treatment for chronic lymphocytic leukemia (CLL) in combination with chlorambucil [33–35]. Thus, using glycoengineering to modulate key enzymes of fucosylation serves as a mature technology to modify antibody therapeutics with acknowledged effectiveness.
In addition to Asn297 afucosylation, a potential application of antibody glycosylation has been discovered in regard to digalactosylated modification as a principal modulator in the selective transfer of antibodies across the placenta. Martinez et al. [36] revealed that the efficiency of antibody transfer across the placenta in human immunodeficiency virus (HIV)-infected women was affected by maternal IgG characteristics, such as binding to placentally expressed 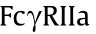 and
and  , as well as Fc region glycan profiles. Furthermore, the bisected and disialylated modification of glycans in the Fc region was associated with transfer efficiency, and a lower fucose level was demonstrated to be a favorable factor in this process. In addition, Jennewein et al. [37] focused on neonatal vaccination and investigated the Fc profiles of neonatal and maternal antibodies. They found that digalactosylated Fc-glycans, which are characterized by selective binding to FcRn and , resulted in preferential antibody transfer to efficiently leverage innate immune cells—mainly NK cells on the very first day of life—and thus contributed to enhanced NK cell degranulation and cytokine secretion [38,39]. These findings provide insights for next-generation maternal vaccines to elicit antibodies for neonate aid.
, as well as Fc region glycan profiles. Furthermore, the bisected and disialylated modification of glycans in the Fc region was associated with transfer efficiency, and a lower fucose level was demonstrated to be a favorable factor in this process. In addition, Jennewein et al. [37] focused on neonatal vaccination and investigated the Fc profiles of neonatal and maternal antibodies. They found that digalactosylated Fc-glycans, which are characterized by selective binding to FcRn and , resulted in preferential antibody transfer to efficiently leverage innate immune cells—mainly NK cells on the very first day of life—and thus contributed to enhanced NK cell degranulation and cytokine secretion [38,39]. These findings provide insights for next-generation maternal vaccines to elicit antibodies for neonate aid.
《3.2. Fc amino acid alterations》
3.2. Fc amino acid alterations
Since sequence variations in the Fc region affect specificities and affinities to FcRs as well as complement protein C1q, modification of this constant region sequence by means of amino acid mutations makes it possible to alter the effector functions. The modification strategy depends on the effects of binding to , in that  ,
, 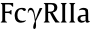 ,
,  , and
, and 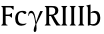 are activating receptors [40], whereas
are activating receptors [40], whereas  is the only one to exert inhibitory effects [21]. Much has been done to regulate FccRIII binding, given its ADCC-initiating role. With the mutations of serine 239 aspartic acid (Ser239Asp)/isoleucine 332 glutamic acid (Ile332Glu)/alanine 330 leucine (Ala330Leu) (referred to as DLE), Herceptin was found to recruit more NK cells, which then killed target cells upon first contact regardless of cancer antigen expression levels [41,42]. The anti-integrin antibody MEDI-522 and Rituximab showed similar improved efficacy with the DLE mutations [43]. Furthermore, asymmetric Fc mutation has inspired researchers to combine distinct mutation formats in one antibody Fc region. The combination of leucine 234 tyrosine (Leu234Tyr), glycine 236 tryptophan (Gly236Trp), and Ser298Ala (referred to as YWA mutations) in one Fc heavy chain was introduced to another heavy chain with DLE mutations, which resulted in more potent ADCC in vitro [44]. As for ADCP by macrophages via binding, Ser239Asp/ Ile332Glu/Gly236Ala was found to promote -dependent ADCP and
is the only one to exert inhibitory effects [21]. Much has been done to regulate FccRIII binding, given its ADCC-initiating role. With the mutations of serine 239 aspartic acid (Ser239Asp)/isoleucine 332 glutamic acid (Ile332Glu)/alanine 330 leucine (Ala330Leu) (referred to as DLE), Herceptin was found to recruit more NK cells, which then killed target cells upon first contact regardless of cancer antigen expression levels [41,42]. The anti-integrin antibody MEDI-522 and Rituximab showed similar improved efficacy with the DLE mutations [43]. Furthermore, asymmetric Fc mutation has inspired researchers to combine distinct mutation formats in one antibody Fc region. The combination of leucine 234 tyrosine (Leu234Tyr), glycine 236 tryptophan (Gly236Trp), and Ser298Ala (referred to as YWA mutations) in one Fc heavy chain was introduced to another heavy chain with DLE mutations, which resulted in more potent ADCC in vitro [44]. As for ADCP by macrophages via binding, Ser239Asp/ Ile332Glu/Gly236Ala was found to promote -dependent ADCP and  -dependent ADCC activity in vitro [45]. Neverthe less, this mutation inevitably led to a 13-fold greater binding affinity to
-dependent ADCC activity in vitro [45]. Neverthe less, this mutation inevitably led to a 13-fold greater binding affinity to  in view of the 90% sequence similarity between and inhibitory [46]. In contrast, Margetuximab targeting human epidermal growth factor 2 (HER2) with the optimized mutations phenylalanine 243 leucine (Phe243Leu)/arginine 292 proline (Arg292Pro)/Tyr300Leu/valine 305 isoleucine (Val305Ile)/Pro396Leu to obtain a reasonable ratio showed better ADCC activity compared with that of Herceptin [47,48].
in view of the 90% sequence similarity between and inhibitory [46]. In contrast, Margetuximab targeting human epidermal growth factor 2 (HER2) with the optimized mutations phenylalanine 243 leucine (Phe243Leu)/arginine 292 proline (Arg292Pro)/Tyr300Leu/valine 305 isoleucine (Val305Ile)/Pro396Leu to obtain a reasonable ratio showed better ADCC activity compared with that of Herceptin [47,48].
When it comes to CDC-related modification, the influence on other effector functions such as ADCC and ADCP should not be neglected, since the triple mutation Ser267Glu, His268Phe, and serine 324 threonine (Ser324Thr) has been found to largely improve CDC at the expense of reduced ADCC and ADCP via increasing the affinity to inhibitory [41]. Therefore, the ratio of the bound activating and inhibitory  should be taken into account in Fc region sequence alteration.
should be taken into account in Fc region sequence alteration.
As for half-life prolongation, the quadruple mutation Ser298Ala, Glu333Ala, lysine 334 alanine (Lys334Ala), and Asn434Ala was found to enhance binding with both FcRn and [49]. The Glu294 deletion resulted in higher sialylation of the Asn297 glycan on the Fc and showed an increased antibody half-life in vivo [50]. It can be concluded that serum half-life regulation is not limited to FcRn binding, but includes sialylation as well.
《3.3. Cross-isotype or cross-subclass exchanges》
3.3. Cross-isotype or cross-subclass exchanges
Considering the binding of different Fc isotypes to their corresponding FcRs in order to induce respective immunological responses, this category of modified antibodies aims to enhance ADCC and/or CDC by recombining and/or replacing sections of natural Fc domains with those from different isotypes/subclasses to form cross-isotype antibodies. Researchers have attempted to engage multiple Fc sections. As neutrophils engage the Fc of immunoglobulin A (IgA) antibodies via the Fc alpha receptor I (FcαRI) [51,52], Chintalacharuvu et al. [53] appended single domains of IgA2 to the end of the 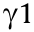 constant region and substituted the CH1 domain of with that of a1 to create a four domain constant region (CH1a–CH2g–CH3g–CH3a) termed IgGA. This cross-isotype antibody was shown to be less affected by pH in comparison with IgG1, and was able to mediate the complement-dependent lysis of sheep red cells. Borrok et al. [54] reported the tandem cross-isotype IgG/IgA, which was created by fusing the hinge, CH2, and CH3 of IgA2 to the C-terminus of IgG1. It was able to bind FcαRI,
constant region and substituted the CH1 domain of with that of a1 to create a four domain constant region (CH1a–CH2g–CH3g–CH3a) termed IgGA. This cross-isotype antibody was shown to be less affected by pH in comparison with IgG1, and was able to mediate the complement-dependent lysis of sheep red cells. Borrok et al. [54] reported the tandem cross-isotype IgG/IgA, which was created by fusing the hinge, CH2, and CH3 of IgA2 to the C-terminus of IgG1. It was able to bind FcαRI,  , and FcRn with the approximate affinities of wild-type IgA and IgG, respectively, in vitro, such that it could mediate ADCC through both neutrophils and NK cells, whereas C1q binding was compromised three-fold in comparison with IgG1.
, and FcRn with the approximate affinities of wild-type IgA and IgG, respectively, in vitro, such that it could mediate ADCC through both neutrophils and NK cells, whereas C1q binding was compromised three-fold in comparison with IgG1.
In view of its high affinity to C1q in vitro, IgG3 was introduced to create IgG1/G3 cross-subclass antibodies with enhanced IgG3 effector functions [55]. The more effective chimeric format is the fusion of CH1 and the hinge region of IgG1 to the IgG3 Fc region named 1133. The CDC activity of 1133 is better than that of either IgG1 or IgG3, even with low antigen levels [55,56]. In addition, subtle modification could be utilized to accomplish gain-of-function in a subclass without the expected properties. IgG2 and IgG4 are incapable of inducing CDC as IgG1 and IgG3 do [57]. In accordance with this strategy, CDC activity can be endowed by replacing IgG2 CH2 domain with that of IgG3. Furthermore, changing the IgG4 residue at position 331, where C1q binds IgG, in order to elicit CDC activity to match IgG1, gave rise to a moderate level of CDC [56]. Hence, residues associated with the specific functions of antibodies can be introduced to obtain gain-of-function in corresponding Fc domains in order to confer effector functions.
《3.4. Antibody–drug conjugates》
3.4. Antibody–drug conjugates
Having evolved from Paul Ehrlich’s ‘‘magic bullet” concept, monoclonal antibodies (mAbs) can be conjugated with various effector molecules, such as cytotoxic agents, radiopharmaceuticals, and immunotoxins, to generate a new pattern of target therapy: ADCs [58]. In general, an ADC consists of three components: the first component is a mAb, which is armed with the second component, effector molecules as the ultratoxic payload (or warhead) to induce the death of target cells, which occurs through the third component, a stable and conditionally biodegradable linker [59]. The availability of ADCs in clinical practice relies on every component having appropriate characteristics, which include strengthened stability of the linker, outstanding potency, sufficient payload release, and—more importantly—humanized or human antibodies as a framework to attenuate immunogenicity and improve selectivity. Progress has been made in this area, with a total of ten ADCs having been approved for clinical application. Moreover, glembatumumab vedotin (CDX-011), which was developed by Celldex/Seattle Genetics to target glycoprotein nonmetastatic melanoma protein B (gpNMB) against advanced melanoma, has demonstrated modest activity and an acceptable safety profile in a phase II study [60], and holds promise to be approved by the FDA as an ADC for clinical practice.
The components of ADCs can be regarded as modification targets for modifying ADCs to promote their therapeutic effects. As the pivotal determinant of ADC efficacy, the cytotoxic payload mainly targets either DNA (e.g., calicheamicins and duocarmycins) or tubulin (e.g., maytansines and auristatins). Szot et al. [61] reported a potent monomethyl auristatin E (MMAE)-linked ADC that elicited anticancer activity through a prodrug activated by stroma cells in a tumor microenvironment (TME), which was termed drug activation and release through stroma (DAaRTS). Tumor-associated stromal cells released active free drug MMAE, killing nearby proliferating tumor cells in a target-independent manner. This design has overcome the problem of conventional ADCs being limited to the treatment of antigen-positive patients.
Loading a large amount of toxins often unexpectedly results in impaired hydrophilicity, which compromises the ADC’s biophysical properties, including decreased solubility, aggregation reactions, and a low drug-to-antibody ratio (DAR) of no more than 3–4 [62–64]. Schneider et al. [59] developed a novel class of hybrid ADC named dextramabs. The therapeutic antibody trastuzumab was equipped with a multivalent dextran polysaccharide, which achieved an elevated DAR, remarkable hydrophilicity, efficient toxic agent load, and high toxicity in vitro, as proved by HER2+ breast cancer cell lines.
With regard to linker modification, as the linker on an ADC can also be associated with detectable labels such as fluorophores [65], methods to label antibodies usually rely on the modification of interchain disulfides by means of genetically encoded amino acids such as free cysteine residues [66]. These conventional methods have the following drawbacks: ① Modification of the cysteine residues after disulfide reduction is difficult and may bring out heterogeneous mixtures [67]; and ② genetically inserted noncanonical amino acids in antibodies could risk altering the native sequence of the antibody and sacrificing binding affinity to the target antigen [68]. Matos et al. [69] developed a region-selective lysine methodology through a core organic chemical reaction in which methyl 2-(sulfonyl methyl) acrylate (abbreviated as 1c) directly modified a single lysine residue on the native protein sequences of the antibody without genetic engineering. The modification of trastuzumab not only achieved site-specific fluorescent labeling, resulting in trastuzumab-1c-fluorescein isothiocyanate (FITC), but also retained the specificity of trastuzumab toward its target antigen (HER2/c-erb-2) at a concentration range identical to that of its unmodified counterpart [70]. This method was further validated for the construction of a stable and functional ADC through the conjugation of the kinase inhibitor crizotinib, which was approved for the treatment of anaplastic lymphoma kinase (ALK)-rearranged non-small-cell lung carcinoma (NSCLC), to the acrylate present in trastuzumab-1c [71]. The ADC trastuzumab1c-crizotinib maintained antigen binding properties, and the secondary structural content retained its specificity toward SKBR3 cells expressing high levels of HER2 antigen.
When modifying antibody drugs by introducing additional elements, including special linkers and effective drugs, the properties of ADCs such as hydrophilicity, half-life, and receptor affinities should be comprehensively considered.
《3.5. scFv for chimeric antigen receptor T cells》
3.5. scFv for chimeric antigen receptor T cells
A CAR is a protein that consists of the scFv from the specific antibody fused with the intracellular signaling transducers, the f subunit of CD3 (CD3ζ) and costimulatory domains such as CD28 or anti-human-tumor necrosis factor receptor superfamily member 9 (4-1BB) [72]. Autologous T cells from patients are genetically engineered to express CARs and are termed as CAR-T cells; they are subsequently infused back into the patients to recognize the specific antigens and kill the antigen-expressing cells, thereby serving as a cellular therapy. CARs can be regarded as a peculiar format of modified antibodies expressed by T cells. It can be inferred that CARs derived from certain antibodies determine the specificity of targets and therapeutic effectiveness. In 2017, Kymriah (tisagenlecleucel-T, CTL019), developed by Novartis Pharmaceuticals Corporation (Switzerland), and Yescarta (axicabtagene ciloleucel), developed by Kite Pharma Inc. (USA), were approved for relapsed or refractory large B cell lymphoma after two or more lines of systemic therapy such as diffuse large B cell lymphoma (DLBCL). As of 31 December 2020, over 900 clinical studies associated with CAR-T have been registered as clinical trials↑ (↑ https://clinicaltrials.gov.), China is the most active area of clinical research (420 items) and has the highest rate of CAR-T research among all clinical trials, followed by the United States (261 items).
Although CAR-T therapy has shown remarkable efficacy [73,74] through the enforcement of the immune system and the redirection of specific immunocytes, this strategy presents three issues:
(1) Safety issues may follow treatment. Caused by the recognition and killing of antigen-expressing cells [75], the most epidemic toxicities brought about by CAR-T are cytokine-release syndrome (CRS) [76] and neurotoxicity reflected by cerebral edema [77]. Lowering the doses of CAR-T cells was shown to decrease toxicities, albeit at the cost of decreasing clinical effectiveness [78]; therefore, IL-6R antibody (Tocilizumab), IL-6 antibody (Siltuximab), Janus kinase (JAK) inhibitors, and corticosteroids have been utilized to block pro-inflammatory IL-6 signaling, in order to reverse fever, hypotension, and hypoxia [79,80]. The mechanisms of both toxicities remain elusive, partially due to a lack of informative animal models for preclinical studies.
(2) The specificity and selection of the target antigen can be problematic. ‘‘On-target, off tumor” toxicity gave rise to unexpected damage to patients, since noncancerous cells expressed the targeted antigens as well [81]. Worse still, antigen escape has been continuously observed in treated patients [82]. Therefore, the discovery of highly cancer-specific antigens is greatly needed to prevent extra events in patients in poor condition. Furthermore, multiple targets including different antigens and spliced variants of single antigens should be covered in CAR-T therapy.
(3) There may be overestimation of autologous T cells efficacy. T cell immunity is regulated by complicated networks in the TME, especially in solid tumors, including but not limited to immune checkpoints such as programmed cell death-ligand 1 (PD-L1) and metabolic alterations such as hypoxia and oxidative stress. As a result, T cell immunity efficacy can be compromised [83]. In addition, immune defects related to cancer treatments worsen the immune status and functions of the patients. Thankfully, genome-editing techniques such as transcription activator-like effector nuclease (TALEN) and clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 have been applied to T cell engineering for the functional enhancement of CAR-T cells [84,85]. Moreover, in ongoing studies, universal CAR-T cells from healthy donors have been shown to potentially overcome the impairment of immune status [84–86] if the inherent barriers of major histocompatibility complex (MHC) are eliminated [87,88]. Thus, much remains to be explored in immunoregulatory networks, optimal antigen selection, and sophisticated affinity modulation, particularly by genetic engineering, for the specific modified antibody format, the CAR.
《3.6. Bispecific antibodies》
3.6. Bispecific antibodies
bsAbs refer to a large family of recombinant molecules that are designed to recognize two disease-related targets or distinct epitopes within one molecule, which breaks up the natural IgG architectural format [89], since inhibiting a single target may fail to achieve significant efficacy. In general, constructive strategies of bsAbs aim to achieve advantages such as desired clinical efficacy, favorable physicochemical properties, minimal or no immunogenicity risk, and scalable manufacturability, even without intellectual property issues. The efforts that have been put into constructive strategy exploration have resulted in around 100 different formats in the bsAb family [89], all of which can essentially be divided into two major categories: those bearing an Fc region and those lacking an Fc region. Among these constructive formats, it is acknowledged that engineered-antibody fragments, such as scFv, the single domain antibodies from Llama (VHH), and Fab, have mostly been used as building modules—that is, as basic blocks to design and construct bsAbs [90–92]. These fragments range from tandem scFv/VHH to fragments-attached IgG-like molecules against anticipated targets [89,93,94]. Here, we describe the potential of bsAb candidates to become the next wave of modified antibody-based therapies, with a main focus on the moieties in clinical development.
To date, over 85 bsAbs are in clinical development, and more than 20 commercialized technology platforms are available for bsAb therapeutics creation and development [89]. Among the developed bsAbs, two have been marketed: blinatumomab (CD3 × CD19, where × denotes a combination of two antigen specificities) [95], a fragment-based bispecific T cell engagers (BiTCEs) for acute lymphatic leukemia (ALL) and B cell-ALL; and emicizumab (coagulation factor IXa (cFIXa) × coagulation factor X (cFX) and/or coagulation factor Xa (cFXa)), a full-size bispecific IgG for the routine prophylaxis of hemophilia A patients.
bsAbs can be categorized according to the engaged cells—that is, as BiTCEs, which activate T cells (CD3+ ) to initiate T cell receptor (TCR) signaling; or as CD16a-associated bsAbs, which activate NK cells (CD16a+ ) and dendritic cells (DCs) to induce ADCC. To date, there are 158 BiTCEs in total, and the targets mainly concentrate on CD19 (13 BiTCEs), epidermal growth factor receptor (EGFR) (12 BiTCEs), HER2 (11 BiTCEs), B cell maturation antigen (BCMA) (10 BiTCEs), and prostate-specific membrane antigen (PSMA) (8 BiTCEs). The redirected optimized cell killing (ROCK) technology platform developed CD16a-engaged bsAb AFM24 (EGFR × CD16a), AFM26 (BCMA × CD16a), and AFM13 (CD30 × CD16a). All of them showed similar affinity to different isotypes of CD16a encoded by CD16- 158V and CD16-158F, which overcame the lower affinity of CD16- 158F to the Fc region. AFM24 and AFM26 were more potent to kill target cells in vitro, while AFM13 was found to have acceptable safety and tolerability, as well as a high overall response rate (ORR) in phase I/II clinical studies
It may also be practical to classify bsAbs by their mechanisms, as follows: ① Combinatorial bsAbs are designed as a mixture of antibodies, which may lessen the patients’ economic burden. However, the ratio of each component, which is fixed in the development procedure, cannot be personalized in accordance with different patients’ conditions. Similarly, there are difficulties in optimizing the pharmacodynamics and tolerability [89]. ② Obligate bsAbs are those in which the bodily linkage of binding domains generally creates a novel functionality; that is, a characteristic which can’t be implemented by using an antibody mixture. Temporal obligate bsAbs mediate the sequential binding of the components. For example, binding the first domain facilitates the second domain’s function by making the distal target accessible. Spatial obligate bsAbs exert functions that are dependent on the simultaneous binding of the domains. Consequently, targeted antigens are redistributed in space and facilitate downstream effects following this bsAb-dominated positioning. The interaction pattern of spatial obligate bsAbs is similar to that between effector cells and antigen presentation cells or enzymes and their substrates. The phases and progress of the bsAb clinical studies registeredy are shown in Fig. 2 according to this classification.
《Fig 2》

Fig 2. An updated summary of the clinical trial stage of bsAbs. Following the categorization reviewed by Labrijn et al. [89], the bsAbs registered are classified according to indications (data was accessed in September 2019). Tumors are the major approved investigational diseases for bsAbs with a proportion of 85.2% (75/88). Among them, 45 bsAbs are for solid tumors and 37 bsAbs are for hematological tumors. It is notable that MGD014 (CD3×HIV-1 Env) for HIV infection and MEDI3902 (pslexopolysaccharide (Psl) ×the V-antigen of Pseudomonas aeruginosa (PcrV)) for the prevention of Pseudomonas aeruginosa pneumonia have sparked hope for the management of refractory infections. Catumaxomab (CD3×epithelial cell adhesion molecule (EpCAM)) was withdrawn in 2017 for commercial reasons and was associated with fatal toxicity at low doses.
The ongoing innovations with the new concepts that are arising may guide future directions in this field. It is attractive that the non-protein delivery of therapeutic bsAbs by mRNA or DNA coding shortens manufacturing duration to attain pharmaceutical-grade nucleic acid products. Stadler et al. [96] described a fragmentbased BiTCE (CD3 × tight-junction protein claudin 6 (CLDN6)) encoded by an optimized, nucleoside-modified mRNA with sustainable production in vivo. Targeting and translation were ensured in the liver after intravenous administration through polymerbased and/or lipid-based formulation. This system was proven to be as effective as the corresponding purified protein-delivered bsAb. With better thermostability, the DNA-encoded format of bsAbs delivery enables less rigorous transfer conditions and relatively long-term storage. Relevant studies have been accomplished: Petal et al. [97] and Digiandomenico et al. [98] demonstrated a DNA-encoded symmetric bsAb format with sustained in vivo expression targeting the pathogenic components such as the V-antigen of Pseudomonas aeruginosa (PcrV) and pslexopolysaccharide (Psl) of Pseudomonas aeruginosa. This creation displayed minor differences in potency compared with the protein-delivered version. Furthermore, Keyt and colleagues made use of a J chain, which affiliated with the natural framework of IgA and IgM, and attached it to the effector cell targeting arms. This innovation was shown to permit bispecific formats with a 1 + 4 design, as in the case of dimeric IgA, or with a 1 + 10 design, as in the case of pentameric IgM, which could induce the targeting of pathogenic drivers with higher avidity being expressed at low levels. Flexible and less constrained structures endow bsAb with incomparable advantages in exploiting functions and promoting efficacy for clinical management. With the prosperity and improvement of commercialized technology platforms, new therapeutic targets should be explored to take advantage of bi-specificity.
《4. Progress in modified therapeutic antibodies in China》
4. Progress in modified therapeutic antibodies in China
China has achieved breakthroughs in the R&D of therapeutic antibodies, based on the introduction and absorption of advanced technology in recent years. Immune checkpoint antibodies for tumors are one example, with 25 therapeutic antibodies having been registered at the Center for Drug Evaluation (CDE), National Medical Products Administration (NMPA) of China. Among these, Toripalimab (JS-001), which was developed by Shanghai Medipharm Biotech Co., Ltd. (China), is the first mAb against programmed cell death receptor-1 (PD-1) that has been conditionally approved by the NMPA of China for the treatment of unresectable or metastatic melanoma in clinical trials in China [99]. In May 2019, Camrelizumab (SHR-1210), a humanized IgG4-κ anti-PD-1 mAb being developed by Jiangsu Hengrui Pharmaceuticals Co., Ltd. (China), received conditional approval in China for the treatment of relapsed or refractory classical Hodgkin’s lymphoma [100]. These domestically developed products represent China’s potential and capability in the innovation and development of biotherapeutics.
In the field of modified therapeutic antibodies, the National Key R&D Program of China called Precision Medical Research initiated projects on modified antibodies and immune cells. These projects align the strategic construction of modified therapeutic antibodies for clinical precision medicine and the establishment of criteria for biologics-based individualized treatment with associated genetic phenotype detection reagents and methods. Therefore, R&D on domestically developed modified therapeutic antibodies has been extensive and prosperous in recent years. The modified therapeutic antibodies shown in Table S1 in Appendix A were developed by Chinese scholars and have been approved by the CDE, NMPA of China for clinical trials
《4.1. Modified glycosylation》
4.1. Modified glycosylation
During the outbreaks of Ebola virus disease (EVD) in 2014– 2015, the cocktail of mAbs called MIL77, which was developed by Beijing Mabworks Biotech Co., Ltd., cured two Ebola virus (EBOV)-infected patients from the United Kingdom and Italy↑ (↑ http://1712130038.pool1-site.make.yun300.cn/product/3.html.). The CHO cells for MIL77 expression were glycoengineered to prevent fucosylation, thereby demonstrating the role of afucosylated modification in efficacious antibody production [101,102]. Furthermore, the efficient construction and manufacturing of MIL77 was performed in a timely manner, within three months. This event and the production of MIL77 displayed China’s speed and competence in the biopharma industry. MIL77 was issued clinical approval by the NMPA in 2017, and a phase I clinical study was carried out.
In regard to cancer-related therapies, Zhang et al. [103] developed Metuzumab (HcHAb18), an affinity optimized and afucosylated human–mouse chimeric IgG1 mAb against CD147 with enhanced ADCC. This achievement was ranked in the ‘‘Top 10 Progress in Chinese Medicine Biotechnology” listed by the China Medical Biotech Association in 2016, and the novel mAb HcHAb18, which is based on Metuximab followed by modifications such as humanization and afucosylation, has been used for NSCLC patients as an independently innovative biologic product. By targeting CD147, which is highly expressed in NSCLC, Metuzumab enhances ADCC and sensitizes tumor cells to chemotherapy in combination with chemotherapeutics [103].
《4.2. Antibody–drug conjugates》
4.2. Antibody–drug conjugates
In early September 2019, OBI Pharma, Inc., a biopharmaceutical company in Taiwan Province, China, announced that the FDA had accepted the investigational new drug (IND) application of their novel first-in-class ADC named OBI-999. With proprietary linker technology maintaining a consistent DAR for cancer treatment, this ADC is based on the targeting of Globo-H, a glycosphingolipid that is highly expressed in up to 15 epithelial cancers. In preclinical studies, OBI-999 triggered apoptosis and suppressed metastasis in breast cancer cells and xenograft models [104]. These findings provided insight into the aberrant glycosylation of tumor cells and the original therapeutic targets of solid tumors, and OBI-99 is currently in phase I/II clinical trials.
As a kind of ADC, radioimmunotherapy (RIT) is a crucial part of targeting therapy, since the therapeutics make use of the guidance of specific parent antibodies to target the cells in foci and exert killing properties by radiolabeling. 131I-labeled mouse/human chimeric mAb chTNT, which was developed by Shanghai Medipharm Biotech Co., Ltd. (China), is a radionuclidesstrengthened ADC that is based on tumor necrosis therapy antibodies targeting all degenerating cells with antigens [105,106]. The 131I-chTNT-mediated RIT of 43 patients exhibited significant therapeutic effects in advanced lung cancer [106]. The 131I Metuximab injection named LicartinTM is an 131I conjugated anti-CD147 antibody Metuximab that was the ADC modified by Zhang et al. [103] and Chen et al. [107]. It demonstrated safety and activity in the treatment of primary hepatocellular carcinoma (HCC) patients. In advanced HCC, LicartinTM is effective in the prevention of post-orthotopic liver transplantation (OLT) tumor recurrence and has shown a significantly decreased rate of tumor recurrence of 30.4%, as well as a 20.6% increased survival rate [108]. Another novel anti-CD147 ADC conjugates a potent cytotoxic drug tubulin inhibitor maytansinoid derivative 1 (DM1) via a non-cleavable thioether linker (SMCC) named HcHAb18-DM1. For NSCLC, disturbed mitotic spindle formation and antiproliferative activity of HcHAb18-DM1 treated groups were observed in comparison with the control in vitro. Meanwhile, decreased tumor weight and volume in A549 xenograft nude mice administered with HcHAb18- DM1 supported the promising role of HcHAb18-DM1 for clinical treatment against CD147-positive NSCLC [109].
《4.3. CAR-T》
4.3. CAR-T
After two CAR-T therapeutics were approved by the FDA, the CDE, NMPA of China started to accept applications for investigation of new drugs (INDs) for CAR-T products at the end of 2017. As of 2019, 32 IND applications have been accepted for CAR-T and 13 of them have been approved for clinical studies in China. In terms of targets, 27 of the 32 applications target CD19, four target BCMA, and one targets glypican 3 (GCP3) for solid tumors. Therefore, new targets may need to be exploited, especially for solid tumors and universal CAR-T.
《4.4. Bispecific antibodies》
4.4. Bispecific antibodies
At present, there are 13 ongoing clinical trials for five bsAbs approved by the NMPA of China. Among them, KN046 (PD-L1× cytotoxic T lymphocyte antigen-4 (CTLA-4)), the first of its class, which was developed by Alphamab Co., Ltd. (China), has shown effective PD-1/PD-L1 and CTLA-4 blockade, with potential immune checkpoint inhibitory and antineoplastic activity. This antibody restores immune function and activates a sustained cytotoxic T lymphocyte (CTL)-mediated immune response against tumor cells. Clinical trials have also been approved for HER2×HER2 bsAb (Alphamab Co., Ltd., China) and epithelial cell adhesion molecule (EpCAM) × CD3 bsAb (Wuhan YZY Biopharma Co., Ltd., China). Hence, the ongoing clinical trials of bsAbs developed by China demonstrate its anticipated capability and potential to design and manufacture multiple functional antibodies in this field.
《5. Prospects and conclusion》
5. Prospects and conclusion
Considering the extraordinary achievements that have been made with therapeutic antibodies in the treatment of cancers, autoimmune diseases, infectious diseases, and many other disorders, we can expect the generation of more effective antibody therapeutics with higher specificity, enhanced efficacy, lower toxicity, and better pharmacokinetic profiles. The modification of therapeutic antibodies represents a promising and irreversible tendency to edit antibodies and overcome potential limitations of candidate molecules in the R&D pipeline. In this article, we described six categories of modified therapeutic antibodies: ① modified glycosylation, ② Fc amino acid alterations, ③ crossisotype or cross-subclass exchanges, ④ ADCs, ⑤ scFv for CAR-T cells, and ⑥ bsAbs. Numerous modified biotherapeutics have been granted market approval and are still in accelerated research or undergoing clinical trials. China has achieved breakthroughs in this field, including the development of therapeutic entities of ADCs, CAR-T products, and the implementation of clinical studies for modified therapeutic antibodies.
The construction and updating of new technology platforms are in progress and are paving the way for innovations in and modifications of therapeutic antibodies. For example, the HexaBody technology provided a generation of therapeutics with enhanced effector functions [110,111]. Heavy-chain antibodies (HCAbs) include the camelid single-domain antibody (sdAb, also known as nanobody) and the variable region of the nurse shark antigen receptor (immunoglobulin novel antigen receptor (IgNAR), also called variable domain of IgNAR (VNAR)) [112,113]. Both of these have been found to have a relatively small molecular weight (about 15 kDa), low immunogenicity, broad antigen spectrum, and high penetration ability [114,115]. The potential translation of nanobodies (for thrombotic thrombocytopenic purpura (TTP)) and VNAR products (for nerve system diseases, autoimmune diseases, etc.) displays the advantages and promising future of therapeutic HCAbs [114–116]. Researchers may achieve a breakthrough in the routine administration of therapeutic antibodies, considering their thermal stability (up to 70 °C) [117], proteolytical stability (in gastric and intestinal juice), and solubility [118]. It is still important to prolong the half-life in circulation and maintain an effective concentration of sdAb in order to avoid excess clearance caused by the low molecular weight. The therapeutic effects of sdAb must be observed and followed up in order to enable global marketing and allow sdAb to serve as a first-line treatment. Furthermore, attaching the polymer polyethylene glycol (PEG)—that is, PEGylation—onto the surface of antibody fragments prolongs the antibody half-life in circulation and reduces immunogenicity. Fc-fusion proteins are derivatives of antibodies that do not necessarily retain the variable region, which is not contained in modified therapeutic antibodies, so that Fc-fusion proteins are not covered [119]. Antibody humanization remains a standard procedure for therapeutic mAbs and has been explored worldwide as an independent process in the R&D of therapeutic antibodies. Thus, it is not summarized as a modification approach in this review.
The major obstacles confronting the R&D of modified antibodies and their corresponding strategies can be grouped into the following four issues. First, more detailed revelations of concrete molecular interactions and subtle regulatory mechanisms are still urgently required, since appropriate therapy targets underpin the design, manufacture, modification, and clinical efficacy of antibody therapy. Second, preclinical studies are required in order to resolve the emerging drug resistance of biologics, including but not limited to ADCs and CAR-T therapy [120–123]. Third, bioinformatics fuels the R&D of modified therapeutic antibodies, and many approaches based on high-throughput screening (e.g., phage display and alanine scanning mutations) have been utilized in antibody modification, such as the Fc region alteration of therapeutic antibodies [49,124–126]. Last but not least, precision medicine could be extended to biopharmaceuticals, which would include the exploration of site-specific targets at the amino acid level [127], and development and validation for the immunogenicity assessment of modified antibody products [128].
In short, the modification of therapeutic antibodies enables the control of diseases that are difficult to address and provides reasonable therapeutic choices by achieving accurate targeting, elevated specificity, the elimination of adverse reactions, and improved efficacy. The enhanced efficacy of modified therapeutic antibodies motivates growth and progress in the biopharma industry, which will yield more effective and accessible therapeutics in the long run.
《Acknowledgments》
Acknowledgments
This study was supported by the National Basic Research Program of China (2015CB553701) and the National High Technology Research and Development Program of China (2019ZX09732001).
《Authors’ contributions》
Authors’ contributions
Ji-Min Dai wrote the original draft, while Xue-Qin Zhang and Jing-Yao Dai were involved in the writing in terms of reviewing and editing. Xiang-Min Yang and Zhi-Nan Chen were involved in the conceptualization, editing, and collection of statistical data, and Zhi-Nan Chen was involved in funding acquisition.
《Compliance with ethics guidelines》
Compliance with ethics guidelines
Ji-Min Dai, Xue-Qin Zhang, Jing-Yao Dai, Xiang-Min Yang, and Zhi-Nan Chen declare that they have no conflict of interest or financial conflicts to disclose.
《Appendix A. Supplementary data》
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.eng.2020.06.030.











 京公网安备 11010502051620号
京公网安备 11010502051620号




