

《1. Introduction》
1. Introduction
Our current understanding of the complex biology of solid organs, such as the liver, has been tremendously advanced by a wide variety of methodological approaches based on cell biology, biochemistry, molecular biology, and histology. Conventional methods such as the morphological assessment of tissue structure by histology and immunohistochemistry have generated valuable knowledge and revealed improved treatment options for various liver diseases. Furthermore, due to recent technological advances, it has been possible to gain deeper insights into physiological and pathological tissue organization, as well as dynamic processes taking place in vivo. These novel methods include single-cell targeted transcriptome analysis, mass spectrometry-based metabolite and protein expression studies, and considerable advances in twoand three-dimensional (2D/3D) imaging. This commentary will briefly review the recent progress that has been made in the field of imaging-based liver research, highlighting the novel possibilities of high-dimensional, multiparametric imaging in two and three dimensions, as well as those of dynamic time-lapse in vitro and in vivo imaging (Fig. 1). We will also highlight some potential applications of these techniques for specific research questions in liver biology and disease.
《Fig. 1》

Fig. 1. State-of-the-art imaging methods for phenotypical and functional organ imaging in liver research. Based on explanted/biopsied tissue and conventional sections, histological multiplexing including mass cytometry imaging and fluorescent multiplexing can generate high-dimensional phenotypical data for histocytometry in 2D spatial resolution. Using cellular tomography after chemical tissue clearing permits the 3D microanatomical description of complex structures within organs and developing organisms. Chemical clearing is based on refractive index matching and the removal of light-diffracting structures and light-absorbing chromophores. Intravital microscopy is suitable to visualize dynamic biological processes, such as cellular infiltration, activation, and interaction, in high spatial and temporal resolution over a time period of up to several hours. Created with BioRender.com.
《2. Multiplex methodologies: Imaging cytometry》
2. Multiplex methodologies: Imaging cytometry
Conventional histology has been used over decades to describe pathological changes to tissues, such as immune cell infiltrates in both clinical and experimental liver samples. In the past, the histological description of tissues was mainly based on conventional overview staining, immunohistochemistry, or immunofluorescence imaging, with only a very limited number of markers that could be addressed simultaneously. In the age of single-cell analyses and the growing understanding of different cell subsets, conventional methods fall short at fully deciphering the complex underlying biology. Therefore, describing the intricate relationships between cell types, effector molecules, and underlying genetic pathways in situ requires an approach that includes multiparametric visualization. A certain number of methods currently allow for fluorescence-based high-plex histology, on either formalin-fixed paraffin-embedded (FFPE) tissues or cryopreserved tissues; each of these methods has advantages and disadvantages that must be considered.
Tyramide signal amplification is a major advance that makes multiplex immunostaining widely possible (generally up to eight antibodies) [1]. The limitations of this system, such as using a limited number of antibodies and losing flexibility on the antibody panel design over the course of a study, can be overcome in immunofluorescent-based multiplex approaches by using cyclic immunostaining and sequential removal of the signal, followed by digital image alignment [2,3]. The removal of fluorescent signals can be achieved either by chemical or photochemical bleaching, effectively destroying the fluorophores, or by chemical stripping of the detection antibodies and therefore ablation of the fluorescent probes. Such methods, however, require additional sample handling that may alter the tissue integrity or target antigenicity; furthermore, they are prone to errors introduced by incomplete signal removal between cycles and the loss or perturbation of antigens during the multistep process. Nonetheless, these issues can be addressed by means of optimized protocols; we have described a multiplex staining protocol that routinely allows 12–16 parameters to be obtained from a single archived paraffin-embedded slide of mouse or human liver (Fig. 2) [2]. The optical resolution limit is mainly based on the imaging system used for data acquisition. Since most approaches rely on conventional wide-field microscopy, the usual resolution limit is 300–500 nm.
《Fig. 2》
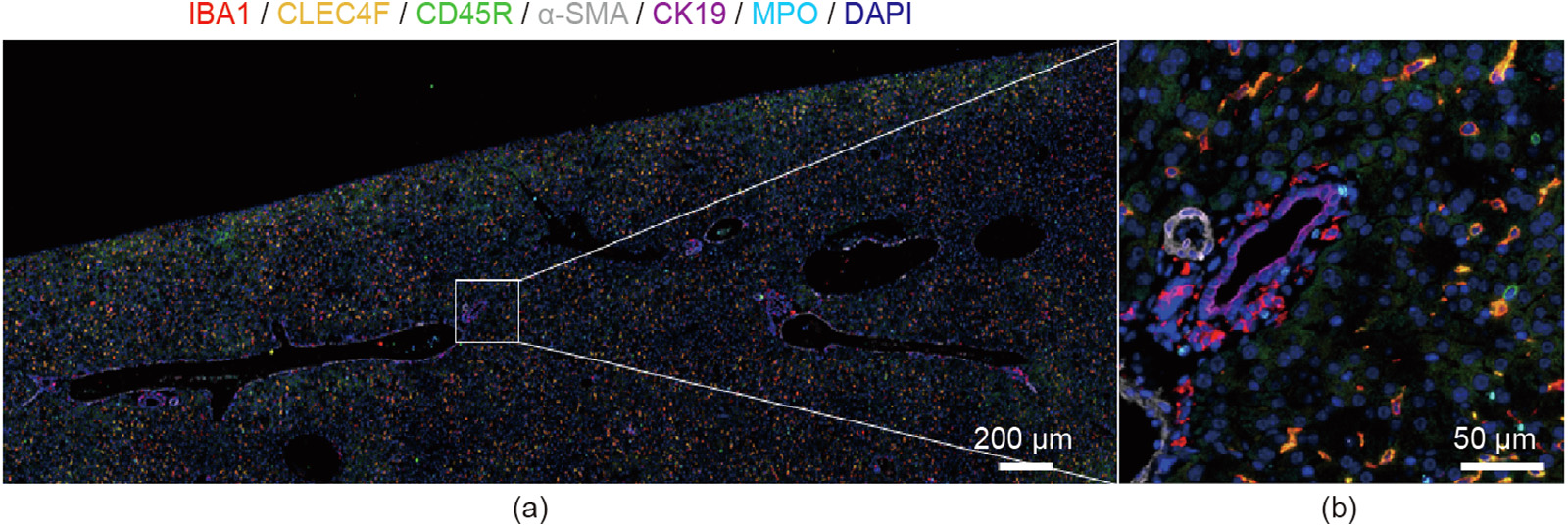
Fig. 2. Representative images of sequential immuno-multiplex fluorescent staining. Sequential immunostaining was performed as previously described [2] on a single healthy control mouse FFPE liver section. (a) A large field of view of the image area; (b) a greater magnification of the white square. Sequential antibody staining and removal allowed the visualization of multiple antigens in a singular tissue section, as indicated by the color legend at the top. IBA1: ionized calcium-binding adaptor molecule 1; CLEC4F: C-type lectin domain family 4, member F; CD45R: cluster of differentiation 45R; α-SMA: α-smooth muscle actin; CK19: cytokeratin 19; MPO: myeloperoxidase; DAPI: 4' ,6-diamidino-2-phenylindole.
Imaging mass cytometry allows the simultaneous labeling of more than 30 parameters, using multifactorial, simultaneous staining with metal isotope-labeled primary antibodies. This method permits the rapid assessment of tissue structure and cell infiltrates from a single tissue section, with very high resolution and complexity; it is similar to high-end flow cytometry or mass cytometry, cytometry by time of flight (CyTOF), and is suitable for unbiased, multifactorial cell cluster data analysis [4]. In imaging mass cytometry, spatial resolution is determined by laser ablation, which is needed to liberate the metal isotopes from the tissue for analysis, and can reach as high as 1 μm.
The major disadvantages of imaging mass cytometry are the need for specialized detection equipment and higher costs due to a lack of standard reagents (Table 1). This technique also requires an individual buildup of an isotope-labeled antibody panel that is applicable for the specific scientific question. Due to the complex process of point by point laser induced ablation followed my mass spectrometric analysis, the effective field of view accessible using imaging mass cytometry is limited to a maximum of only a few square millimeters. In contrast, fluorescence-based image multiplexing uses standard polyclonal or monoclonal antibodies that are widely available, either as directly conjugated reagents or in combination with secondary antibodies. Data acquisition in a multiplex setting can be performed using standard fluorescence microscopes for most research questions; therefore, this method is readily available at most research institutes.
《Table 1》
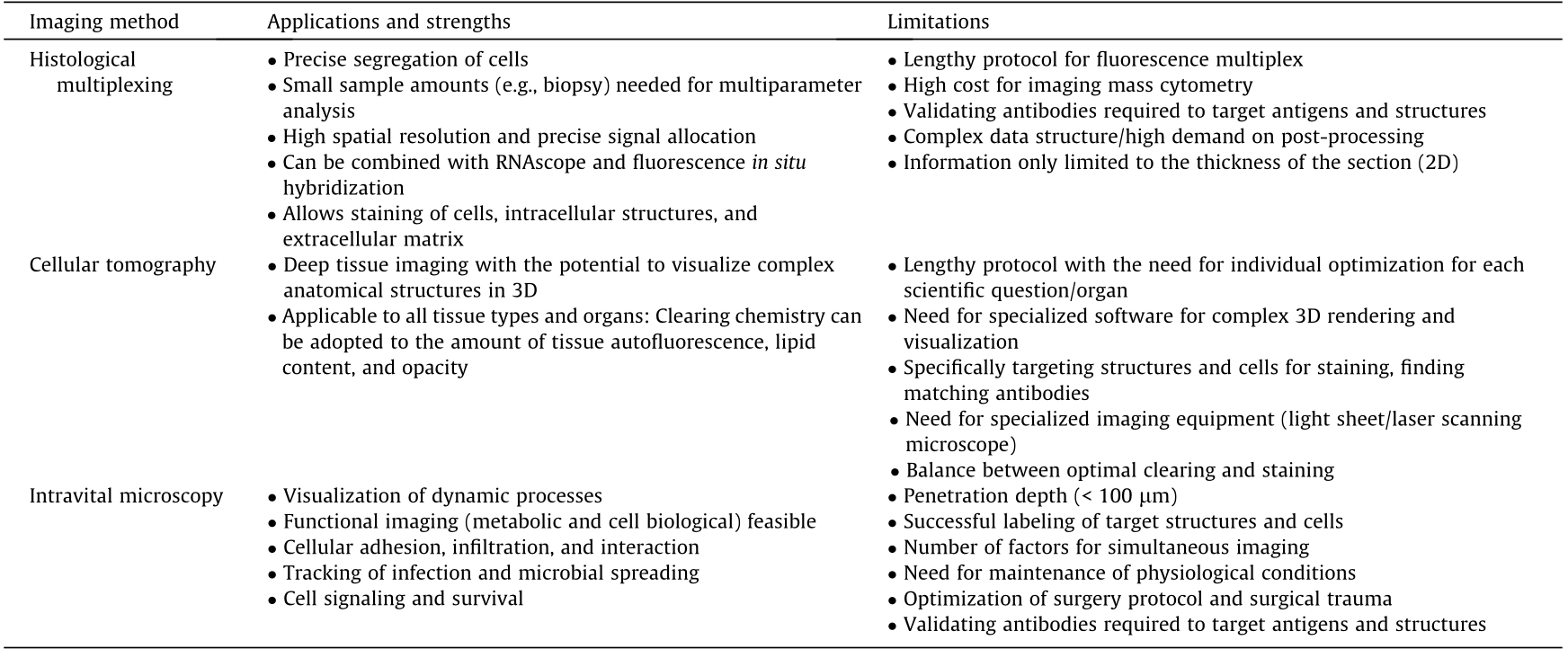
Table 1 Overview of different imaging platforms.
《3. Seeing clearly: The benefits of chemical tissue clearing and 3D tissue tomography》
3. Seeing clearly: The benefits of chemical tissue clearing and 3D tissue tomography
The use of 2D techniques, such as conventional section-based histology, is often insufficient to visualize the complex anatomical structures and spatial correlations within organs. This increases the likelihood of missing localized pathological events, such as the formation of small tumor nodes, focal inflammation, local infection, or—on a larger scale—disorders such as malformations in the vasculature or in the tissue structure itself. The chemical clearing of tissue in combination with high-resolution 3D tomographic imaging using light-sheet, confocal, or multiphoton microscopy circumvents these limitations (Table 1). Tissue clearing is a multistep process that follows fixation of the tissue, based on the stepwise removal of light-absorbing chromophores, scattering surfaces such as lipid membranes, and refractive index matching. Tissue clearing results in translucent tissue whole mounts with a pellucidity of up to several millimeters, depending on the clearing agent [5]. The right choice of clearing procedure and the need to contrast and stain the target structures pose the biggest difficulties in 3D cellular tomography. While solvent-based clearing methods such as benzyl alcohol/benzyl benzoate (BABB) or iDisco rapidly and efficiently clear virtually all different kinds of tissue, they also lead to substantial morphological alterations due to tissue dehydration, and can disrupt the target protein structure that is needed for specific staining using fluorescently labeled antibodies or nanobodies [6]. Furthermore, alcohols often disrupt fluorescent proteins, leading to fluorescence quenching and partial or complete loss of signal. Aqueous-based clearing methods such as Scale, Clarity, or clear, unobstructed brain imaging cocktails and computational analysis (CUBIC) generally take longer to reach the same level of translucency; however, in general, they have been shown to better preserve the structural integrity of the tissue as well as the endogenous fluorescence of reporter proteins such as greenfluorescent protein (GFP), red fluorescent protein, and others (Fig. 3) [7]. Established staining procedures mostly rely on the antibody- or nanobody-based targeting of protein structures, either by the soaking of antibodies into tissues or by the injection of fluorescently labeled antibodies in vivo. Optical resolution strongly depends on the imaging system used to obtain the microscopical data. While light-sheet microscopes generally allow for a higher isotropy of visualizing fluorescence in deeper tissue layers, the range of optical resolution is typically 5–10 μm. Laser scanning systems (both single-photon confocal and multiphoton) provide better optical resolution, with a maximum range of 200–500 nm; however, they usually show a significant performance drop at penetration depths beyond 500–700 μm into the tissue, with a need for data post-processing and normalization.
《Fig. 3》
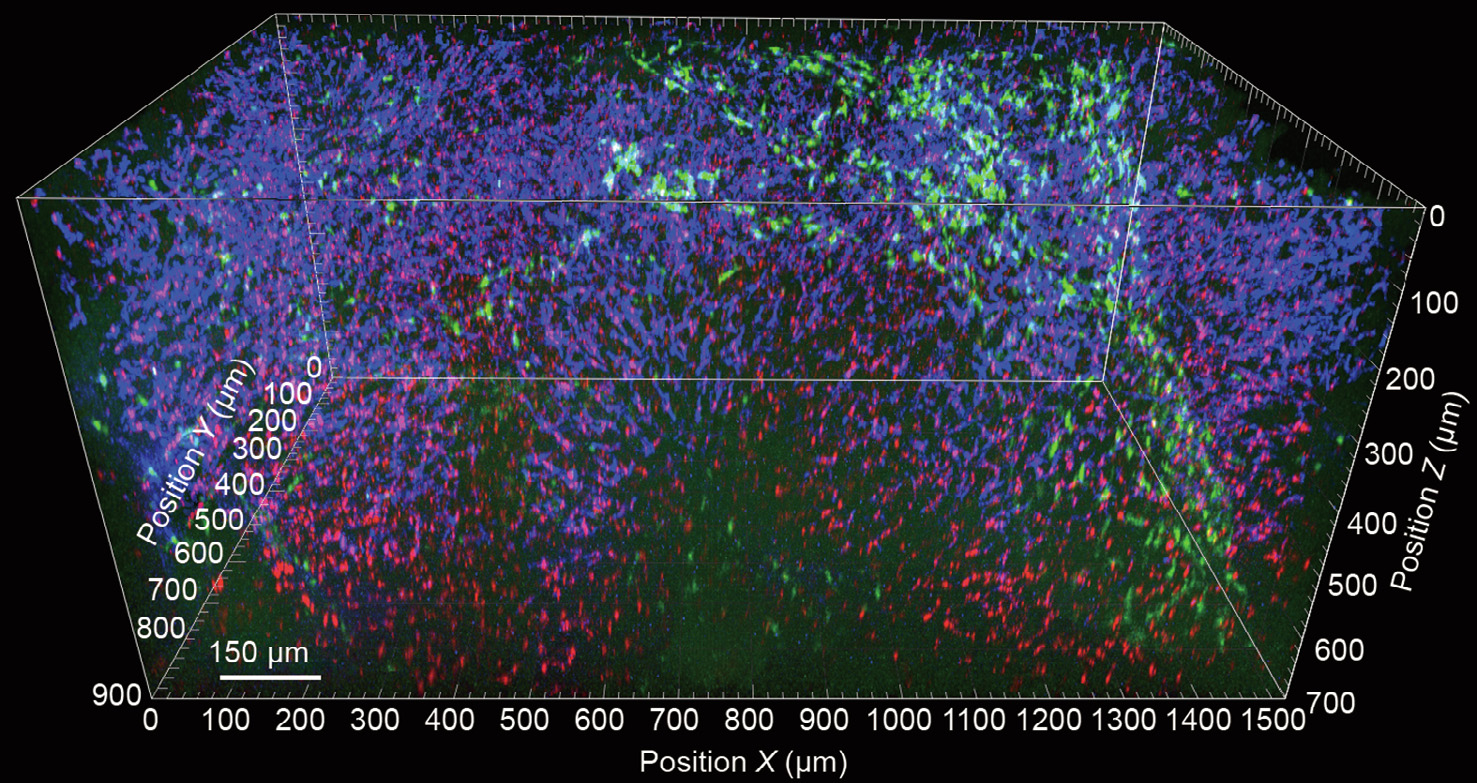
Fig. 3. 3D reconstruction of liver tissue following ex vivo tissue clearing. Liver tissue was isolated from a healthy C–X3–C motif chemokine receptor 1 (CX3CR1).gfp mouse following the intravital injection of F4/80 antibody (blue) and 0.5 μm fluorescent microspheres (red) 30 min prior to tissue explantation. Samples were fixed in 4% formalin solution and subjected to CUBIC-mediated tissue clearing immediately afterwards. Images were obtained using a LaVision Trimscope I multiphoton microscope.
Among the different methodologies, several studies [5,7] and work within our group have indicated that CUBIC may have the highest potential for liver 3D tomography due to its superior preservation of molecular structures and high translucency, despite the high initial chromophore content within the tissue (Fig. 3). Another intriguing possibility is the use of stainless structural imaging based on differential levels of autofluorescence within the tissue. In combination with advanced imaging techniques such as fluorescence lifetime imaging (FLIM), spectral microscopy, or second-/third-harmonic visualization using multiphoton microscopy, these techniques offer novel possibilities for the visualization of tissue structures in homeostasis and disease.
Chemical modification of tissues to enhance the visualization of target structures can also be used to overcome the physical resolution limits described by Ernst Abbe in the 19th century based on the optical diffraction of any given structure. Expansion microscopy has been introduced to the imaging field as one of the most recent technological advances. Instead of enhancing the optical resolution of the imaging system, as performed by the various approaches of super-resolution microscopy (stochastic optical reconstruction microscopy (STORM), stimulated emission depletion (STED) microscopy, structured illumination microscopy (SIM), etc.), expansion microscopy is based on controlled, isometric chemical swelling of the sample in combination with fluorescence staining. In short, samples are prepared by incubation with polymer-forming agents such as acrylamide or sodium acrylate, which results in polymerization and therefore swelling of the sample. This process increases the distance between target molecules, making it possible to improve the spatial resolution of the sample structures by up to four-fold compared with their native state [8].
《4. Intravital microscopy: Adding dynamic changes to spatial information》
4. Intravital microscopy: Adding dynamic changes to spatial information
While time-lapse microscopy has been a tool commonly used in vitro over decades, in vivo imaging has only become available to a wider community within the last two decades. Imaging pathophysiological processes, infections, and immune responses over an extended period of time make it possible to better understand the sequence of events involved in pathophysiological alterations, as well as those in steady-state functions such as metabolism and homeostasis upkeep. Regarding liver imaging, we and others have specifically developed several approaches for short- and long-term intravital liver imaging in rodent models [9–11]. Although the most common applications nowadays are based on the need to image inflammatory processes following infection or tissue damage, intravital microscopy can also be used to image organ functions such as cellular metabolism—for example, the metabolic activity of hepatocytes [12]. This can be achieved by means of FLIM-based visualization of the redox state of metabolites such as nicotinamide adenine dinucleotide/nicotinamide adenine dinucleotide phosphate (NADH/NADPH) or flavin adenine dinucleotide (FAD), making use of the different auto-fluorescent lifetimes depending on their oxidation or reduction. Other functions such as biliary excretion can be immediately assessed by the systemic injection of fluorescent bile acids [13], while qualitative and quantitative blood flow measurements, or visualizing capillary perfusion, can be performed by tracking fluorescently labeled erythrocytes or larger particles injected into circulation. Furthermore, the use of third harmonics allows the visualization of erythrocytes and thus makes it possible to quantify perfusion without the need for an external label.
By specifically making use of the dynamic aspect of intravital imaging, a major application is the measurement of cellular recruitment and cell–cell contacts under real-life conditions (Table 1). For example, even under steady-state conditions, the liver is constantly interrogating and educating bypassing lymphocytes, which is a vital mechanism for the upkeep of peripheral immune tolerance [14]. Intravital microscopy thus provides a unique opportunity to study the cascade of events in the recruitment of different leukocyte cell types, positioning and movement within the tissue, and cellular interactions (Fig. 4). Regarding infectious diseases, intravital microscopy can be used to decipher microbial tissue homing, catching by leukocytes, survival, and proliferation, thereby providing valuable insights into the host–microbe interplay when battling infectious diseases [10]. Direct visualization of viral infection remains challenging at present, due to the small size of the virion particles, which are thus far below the resolution of current optical systems. However, by using genetically modified viruses carrying reporter genes—for example, for GFP—it is at least possible to visualize infected cells in contrast to non-infected cells and study their interplay with the subsequently occurring immune response triggered by the infection.
《Fig. 4》

Fig. 4. Spectral intravital imaging of four-color fluorescent reporter mice with additional antibody staining. CX3CR1-ER-cre × R26_Brainbow2.1 mice carrying cyan fluorescent protein (CFP), GFP, yellow fluorescent protein, and red fluorescent protein were anesthetized and subjected to intravital microscopy of the liver (Zeiss LSM 980 two-photon laser scanning microscopy with spectral detection). Additional antibodies against various macrophage markers (epidermal growth factor-like module-containing mucin-like hormone receptor-like 1 (F4/80), T-cell immunoglobulin- and mucin-domain-containing molecule (Tim4), major histocompatibility complex (MHC)-II, and cluster of differentiation 11b (CD11b)) were injected into the mice to label liver macrophage subsets. (a) Antibody staining; (b) fluorescent proteins; (c) merged image.
Despite the clear benefits of live intravital imaging, several obstacles and limitations must be taken into account (Table 1). Due to the opaque nature of solid tissues, especially chromophore-rich organs like the liver, the penetration of light into and out of the tissue is confined to a very limited depth and reaches only the first 50–100 μm below the surface, even with infrared systems such as multiphoton microscopes. Physically stabilizing the organ is essential for imaging quality and remains challenging due to the anatomical location of the liver, as it resides between the diaphragm and the intestine, with both organs introducing severe motion artifacts.
Similar to the challenges seen in optical clearing, highlighting structures or cells of interest can also pose a potential limitation, giving rise to the need for specific tagging, either by the expression of fluorescent proteins or with fluorescent-labeled antibodies, which do not effectively target antigens buried deep within the tissue further away from the circulation. Also, given the need for continuous sampling over longer periods of time, photobleaching and phototoxic effects must be considered, as they can significantly alter the outcome of the experiment due to alterations of the underlying tissue biology. Similarly, for intravital microscopy the liver and most other organs, it is necessary to surgically expose the organ to make it accessible for imaging. The combined effects of anesthesia, surgical trauma, and the somewhat unphysiological environmental conditions during the imaging process likely also impact the long-term outcome of the imaging results. All these obstacles affect the survival of the animal, currently limiting the time of reliable image acquisition to a period of up to 8 h [9]. An interesting option for circumventing some of the issues related to immediate surgical exposure of the organ is the novel concept of the long-term implantation of abdominal windows, which provides stable access to various abdominal organs such as the spleen, gut, kidney, and liver for up to several weeks [15,16]. In brain research, cranial window implantation has been used extensively over the last years to study neuronal functions using intravital imaging. Pilot studies have also revealed the great potential of long-term visceral imaging, despite caveats such as excessive deposition of extracellular matrix material or chronic inflammation. These preliminary studies have shown, in principle, the possibility of repetitively imaging slow biological processes such as chronic disease progression, wound healing, angiogenesis, or tumor growth.
Current systems using fast FLIM or spectral fluorescence detection, or near-super-resolution detection systems using array detectors for optimized temporal and optical performance, will give rise to yet unprecedented possibilities for overcoming these limitations, and will lead to novel insights into complex in vivo processes in the near future.
《5. Data analysis: New challenges with recent advances》
5. Data analysis: New challenges with recent advances
All of the abovementioned methodological breakthroughs in imaging generate large amounts of data, which poses practical challenges for data storage, processing, and analysis. In particular, challenges arise when it comes to qualitative and quantitative analysis of the data obtained by novel imaging approaches. Feature-based machine learning, dimensional reduction, image segmentation, unbiased cluster analysis, spatial reconstruction, segmentation, and visualization are some of the digital tools under current investigation for data processing [2,3,6]. Recent works have also reported on the rapidly evolving applications of deep-learning- and artificial-intelligence-based approaches to process the ever-growing amount of information gathered from clinical or experimental samples [17].
Despite the power of deep learning approaches in general, the possible uses of cases in the field of scientific image analysis are still relatively limited, due to the heterogeneity of available training data and the lack of specific coherent classifiers to educate neuronal networks. Several open-source platforms, such as PyTorch and TensorFlow, as well as commercially available tools, such as Aivia, have been applied to approach the growing need for artificial-intelligence-assisted image analysis. Yet thus far, the high requirements regarding information technology skills, lack of appropriate training datasets, and individualized questions connected to the immediate scientific question are the main difficulties in implementing deep learning into routine work pipelines for microscopy image analysis.
The more commonly widespread and—to date—more successful approach is user-guided machine-learning-based image segmentation. Several freeware and commercial tools exist (e.g., Ilastik, Weka, APEER, and Intellisys) [18,19], most of which are based on random forest decision trees generated by user-interfacegenerated input data, resulting in prediction maps that are suitable for image segmentation and subsequent use on large image stacks of the same kind.
Digital workflow development and the implementation of novel imaging techniques require specifically trained personnel and often need specialized equipment. The image processing of large, complex datasets relies on sufficient infrastructure with regards to computational power and suitable software solutions; it may also require the individual development of custom solutions, which is very much in parallel with recent advances in the field of single-cell analysis. Although these investments are significant, they are warranted in order to account for the complexity of biology, and will be largely compensated for by the amount of information they will generate, leading to scientific advancements that would otherwise remain inaccessible.











 京公网安备 11010502051620号
京公网安备 11010502051620号




