

《1. Introduction》
1. Introduction
Cement is the most commonly used binder in the construction industry, and it seems that no alternative material can replace it anytime sooner. Among the cement hydration products, calcium silicate hydrate (C–S–H) is the major component and it is responsible for the mechanical properties and durability performance of hardened cement and in turn concrete. The improvement in the properties of the cement matrix requires a deep understanding of the cementitious system at the fundamental scale. Therefore, it is anticipated that the transition from macroscopic properties to ‘‘atomic-scale behavior” of cement matrix may unlock useful information that had been hidden for a long time [1]. The molecular simulations of cement-based materials at the atomic level will eventually allow engineers to design cement mixtures in a way to ensure optimum mechanical properties and durability leading to environmental and sustainable development.
Concrete is a nano-structured, multi-phase, and multi-scale material (i.e., its size varies from nano- to micro- to macrometer), as shown in Fig. 1. It has crystalline and amorphous phases as well as interphase boundaries. Generally, the characteristics at the nano-scale govern the properties at the macro-scale and consequently the macroscopic performance of concrete under working loads [2]. The most critical issue to control most of the properties of concrete is the understanding of the chemistry of cement paste starting from hydration and its related physical changes, such as setting and hardening. Hydration of cement is a complex chemical process involving dissolution, diffusion, nucleation, and growth, and it is associated with both the chemical and physicomechanical changes of the system [3–7]. Such a process is far from fully comprehended since they may undergo complex interactions in a non-monotonic way [8]. As a result, most of the experimental methods that have been used in studying the kinetics of cement hydration are based on monitoring the net rate of the microstructure formation [9]. Nevertheless, none of the experiments provide a rigorous understanding of the hydration mechanism, rather they are effective in comparing and characterizing the affecting parameters, such as water/cement (w/c) ratio, cement composition, temperature, humidity, and so forth. Therefore, there is a need to understand the hydration mechanisms at a nano-scale to achieve the best possible properties from this cheap and commonly used material.
《Fig. 1》

Fig. 1. Schematic of multiscale description of a cementitious system. DEM: discrete element method; FEM: finite element method; XFEM: extended finite element method; XRD: X-ray diffraction; AFM: atomic force microscope; SEM: scanning electron microscopy; TEM: transmission electron microscopes; ITZ: interfacial transition zone.
Since the development of molecular dynamics (MD) simulation in the early 1950s and the evolution of computational power, some breakthrough has been reported on the simulation of structure and performance parameters of the construction materials. In addition to cement, MD modeling has also been used to study other civil engineering materials, including clay [10–12], asphalt [13,14], wood [15–17], and so forth.
With the unlimited increase in computational capacities, such as the use of super-computers, it is now appropriate to assess the possibility of using nano-engineering through molecular simulations in the construction industry. However, to date, there is a lack of a detailed review on the use of molecular simulation to study the interactions of cementitious materials. Consequently, this review intends to fill this gap in the knowledge by providing a comprehensive survey of the extent and level of the current research that has been conducted in this area and scientifically discusses the key results and findings along with providing recommendations for future research.
This review, firstly, provides a brief overview of atomistic modeling and simulation and, secondly, it discusses new directions on simulations of cementitious systems (as presented in Fig. 2) and also the properties of cement are discussed with emphasis on the structure–property relationship. The interaction of cement with water and ions from fundamental bottom-up perspectives is also discussed. It is hoped that the discussion provided in this paper will inspire researchers to predict the macro-level behavior of cementitious materials using molecular-level simulations.
《Fig. 2》
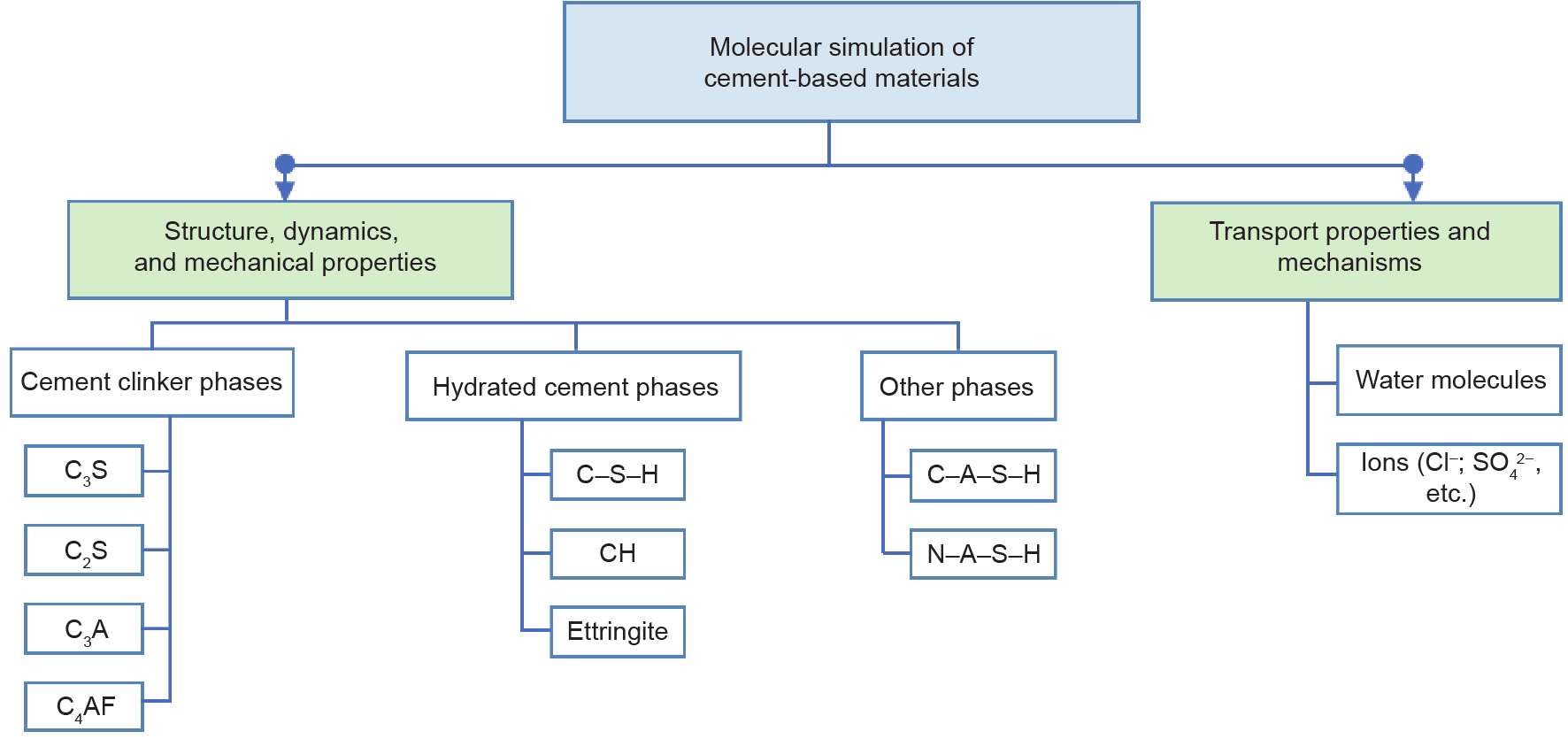
Fig. 2. Molecular simulation of cement-based materials. The terms, C, S, A, and F represent CaO, SiO2, Al2O3, and Fe2O3, respectively, in cement chemistry.
《2. Molecular simulation》
2. Molecular simulation
The integration of the theoretical methods with computational techniques has facilitated the molecular simulation of materials. Molecular modeling becomes an indispensable tool and it has been broadly used in several fields, including chemistry, materials science, pharmacology, biology, energy, and so forth. In recent years, the utilization of molecular modeling in cementitious materials has gained more attention due to the recent advances that have been devoted to nano-engineering and modern materials in the construction industry. In keeping with this trend, molecular simulations offer unique and useful aspects, such as a better understanding of the materials and their interactions at the fundamental scale and testing the effects of independent factors on the behavior of materials [18].
Molecular modeling is based on two main methods: quantum mechanics (QM) and classical mechanics (CM). In the QM approach, Schrödinger’s equation is solved by using either abinitio or semi-empirical methods. QM considers the electronic state of the system, and thus it is computationally expensive and rigorous. On the other hand, numerical modeling using CM relies on classical physics (i.e., non-quantum and based on classical Newtonian mechanics). It treats atoms as mass particles with bonds represented as springs. As a result, these interatomic bonds can be modeled to stretch, bend, and twist (Figs. S1 and S2 in Appendix A).
Classical molecular modeling involves three main techniques: molecular mechanics (MM), MD, and Monte Carlo (MC) simulations (Table S1 in Appendix A). The MM technique relies on traditional CM to model a molecular system. In this method, the interatomic interactions are obtained based on potential functions or commonly called force fields. MC simulations are based on a random sampling of the adsorption process [19].
《3. Molecular simulation of Portland cement phases》
3. Molecular simulation of Portland cement phases
《3.1. Anhydrate phases》
3.1. Anhydrate phases
Ordinary Portland cement (OPC) is a lime-based material. It basically consists of 60%–67% CaO, 17%–25% SiO2, 3%–8% A12O3, 0.5%–6% Fe2O3, 0.5%–4% MgO, 0.3%–1.2% alkalis, and 2%–3.5% SO3 [20]. However, it is common to represent the cement composition in terms of mineralogical composition, namely tricalcium silicate (C3S), dicalcium silicate (C2S), tricalcium aluminate (C3A), and tetracalcium aluminoferrite (C4AF); these are sometimes referred to as alite, belite, aluminate, and ferrite, respectively, due to their similarity with the corresponding minerals. The terms, C, S, A, and F represent CaO, SiO2, Al2O3, and Fe2O3, respectively, in cement chemistry. About 4%–5% gypsum (CaSO4) is generally added to the crushed clinker to regulate the rate of setting of cement [21]. Fig. 3 [22] shows the structure of the four mineral compounds in cement clinker.
《Fig. 3》
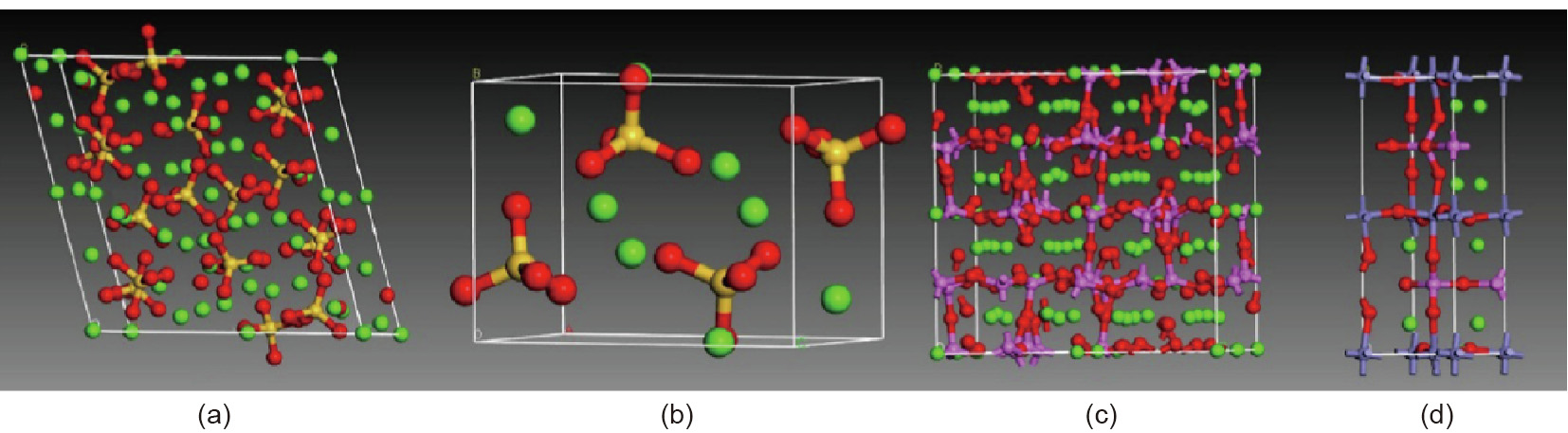
Fig. 3. Structure of major compounds in cement clinker: (a) tricalcium silicate (C3S), (b) dicalcium silicate (C2S), (c) tricalcium aluminate (C3A), and (d) tetracalcium aluminoferrite (C4AF). Reproduced from Ref. [22] with permission.
Molecular simulations have been used to investigate the structure and properties of cement phases to improve the efficiency of cement manufacture and/or to produce the desired type of cement. The MD simulation was also performed through a reactive force field (ReaxFF) to investigate the surface properties and hydration of C3S [23]. It was reported that the static properties, such as surface energy and water adsorption energy, did not provide useful data on the hydration and dissolution, thereby, they cannot be used to predict the hydration mechanism of C3S. In a study by Mishra et al. [24], several force fields, including COMPASS, consistent valence force field (CVFF), and polymer consistent force field (PCFF), were parameterized to study the initial hydration and cohesive properties of C3S. Wu et al. [25] confirmed the suitability of COMPASS force field for evaluating the mechanical properties of silicate phases (C3S and C2S); whereby results comparable with the experimental data were obtained with this simulation. Also, the effect of supercell size on the mechanical properties of cement was reported to be insignificant [25]. Density-functional theory (DFT) approach was also used along with ReaxFF to study the hydration of C2S [26]. Besides, Tao et al. [27] employed ab-initio calculations to investigate the doping behavior of manganese into the cement clinker. They reported that the first principle method provided a fundamental perspective of the experimental results.
Several investigators also modeled the effects of chemical impurities on the structural properties and hydration mechanism of clinker phases. Such modeling would yield low carbon emissions by optimizing cement production. Huang et al. [28] investigated the effect of impurities on the hydration reactivity of C3S using DFT and MD simulations. The atomistic simulations revealed a relation between the long-term hydration and the electronic structure of Portland cement. In addition, Manzano et al. [29] employed the force field-based method and DFT approach to study the effect of chemical impurities, such as Mg2+, Al3+, and Fe3+, on the performance of major clinker phases, namely alite and belite. It was reported that there is a possibility to increase the reactivity of belite that would eventually help in decreasing the energy consumption in cement production. Further, the hydration and dissolution properties of C3S in sodium sulfate solution were also studied using MD simulations [30]. It was reported that the presence of sodium and sulfate ions inhibits the dissolution of ions of C3S. DFT was adopted in studying the adsorption of a water molecule on the surface of C3S. The study is considered a step towards a better understanding of the cement hydration mechanism [31]. The DFT results indicated that dissociative adsorption is more favorable than molecular adsorption, where the former promotes the dissolution process of the cement.
Recently, Sarkar and Mitra [32] investigated the behavior of C3A under compression at the molecular level using the INTERFACE force field (IFF). The unit cell of C3A was made with 72 Ca, 48 Al, and 144 O atoms, where Al and O atoms are covalently bonded in puckered ring. The model was validated with the available experimental and DFT outcomes. It was reported that the energy due to bond length affects the behavior of the material in both the uniaxial and triaxial compression, while the energy due to angle deformation affects the uniaxial compression response only. The edge dislocation of dicalcium silicate was also investigated for the first time by Shahsavari et al. [33]. It was reported that the 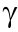 -C2S and α-C2S crystals are the most favorable polymorphs in belite for dislocations, and such a finding may be useful in comprehending the deformation mechanisms in cement clinker.
-C2S and α-C2S crystals are the most favorable polymorphs in belite for dislocations, and such a finding may be useful in comprehending the deformation mechanisms in cement clinker.
An attempt at a molecular dynamic simulation of single crystal gypsum under tensile loading was made by Sarkar and Mitra [34]. The uniaxial and triaxial stress–strain responses were discussed in conjunction with changes in the molecular structure along with interlayer and intralayer separation distances and layer slippages. It was reported that gypsum under tensile loading behaved in an anisotropic way, and this could be explained through the layered structure and the presence of water in between the layers.
《3.2. Hydrated phases》
3.2. Hydrated phases
Cement as a hydraulic binder reacts quickly with water to form new products through the hydration process, which obeys the laws of thermodynamics and kinetics [3]. As mentioned earlier, the hydration process is one of the complex phenomena, and research in this area is still ongoing. In principle, the hydration of silicate phases, mainly C3S, takes place in three main simultaneous reactions according to Eqs. (1)–(3) [35].


In general, the products of cement hydration can be classified into four phases: ① C–S–H, ② calcium hydroxide (CH), ③ ettringite (calcium trisulfoaluminate hydrate, Aft), and ④ monosulfo aluminate (AFm). In the following sub-sections, the molecular simulation of the main hydration products (C–S–H and CH) is discussed.
3.2.1. Calcium silicate hydrate
C–S–H occupies 50%–65% by volume of cement paste. Many researchers considered the models of C–S–H structure either colloidal or layered [36,37]; however, there is a new rigorous model that is yet to be developed. Indeed, the reasons behind this are the complexity in the composition and structure of this basic building block of hydrated cement. However, the integration between the descriptive approaches (based on the experimental evidence) and predictive models (based on numerical simulations) would lead to a realistic model of C–S–H [38]. Fig. 4 demonstrates the history of the development of the C–S–H models, and more details can be found in Refs. [39–56]. The unit cell of C–S–H is composed of calcium silicate sheets, connected by the stable ionic– covalent Ca–O and Si–O bonds with confined water molecules, as depicted in Fig. 5 [57].
《Fig. 4》
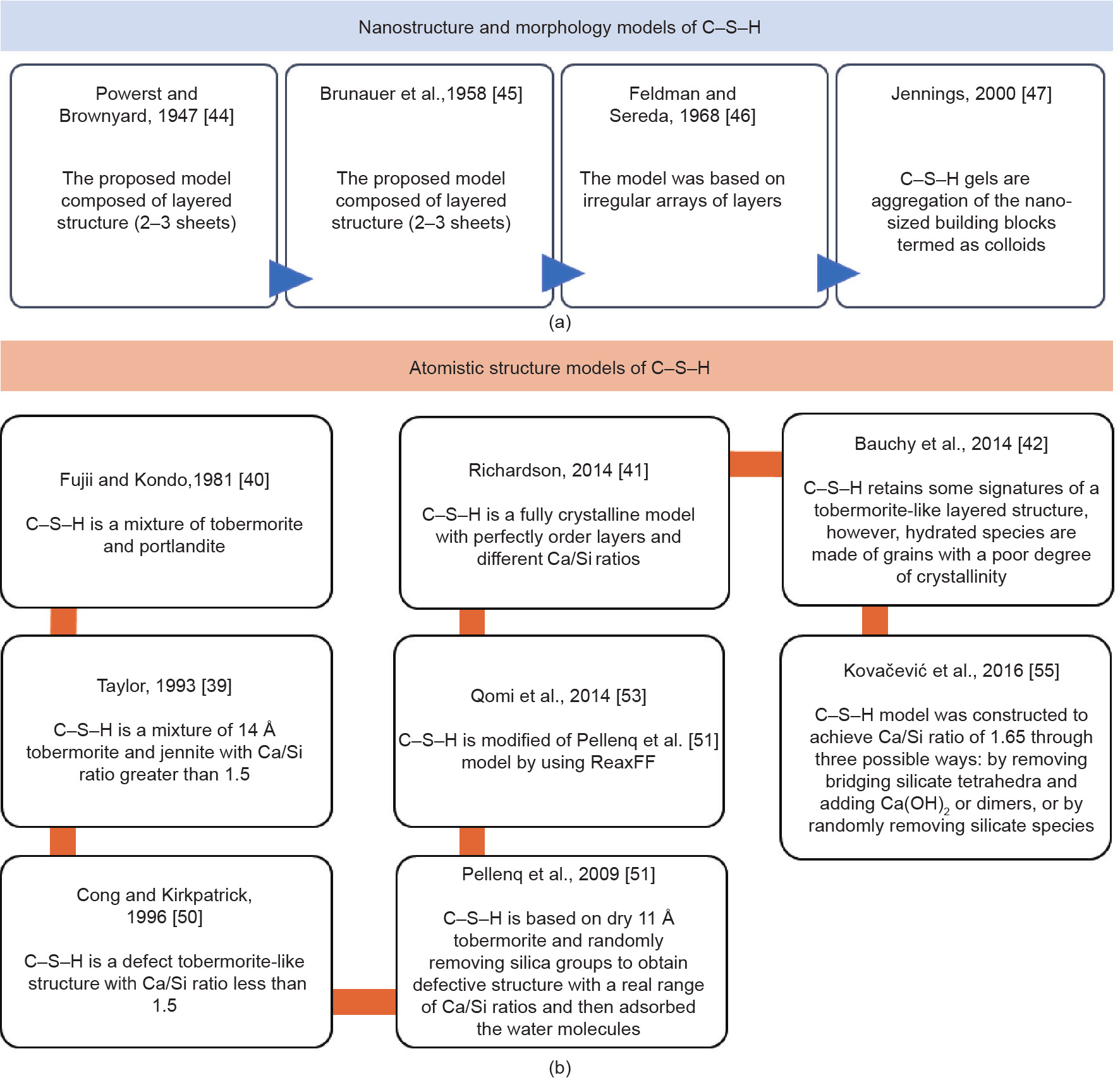
Fig. 4. Morphological models of C–S–H based on experiments; and atomistic structure models of C–S–H based on molecular simulation.
《Fig. 5》
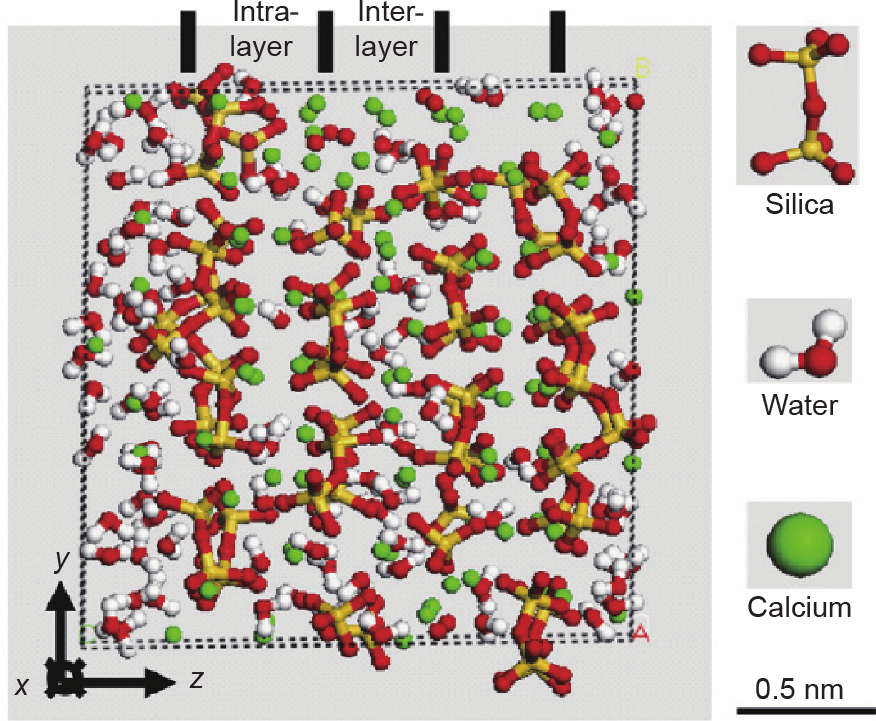
Fig. 5. C–S–H unit cell with highlighted two layers of calcium- and silicon-rich regions (intralayer) separated by water-rich regions (interlayer). Reproduced from Ref. [57] with permission.
Pellenq et al. [51] depicted a molecular model of C–S–H, and it was designated as a ‘‘realistic model.” The model was constructed based on the chemical specificity of C–S–H, and it was modeled using MC simulations. The Ca/Si ratio, density, and mechanical properties of the model were validated using experimental techniques, such as extended X-ray absorption fine structure (EXAFS), X-ray diffraction (XRD), and nanoindentation. Murray et al. [58] presented the monoclinic crystal system of C–S–H in terms of orthogonal coordinates. Therefore, two atomic structures of C–S– H were used. The crystalline structure (based on the Hamid model [52]) and the proposed structure with dimmer silicate chains instead of continuous sheets. The effect of the medium-range Si– O and Ca–O environments of C–S–H was also investigated. Unfortunately, the existing mineral analog-based C–S–H models are not in the line with the experimental results of cementitious C– S–H. Further, they were not able to explain the behavior at the atomic scale. Therefore, some researchers combined the DFT and MD methods to propose a new model. Manzano et al. [59] used ab-initio calculations to identify the structure of C–S–H.
The effect of Ca/Si ratio on the molecular structure and mechanical properties of C–S–H using MD simulation was studied in detail. Using the small-angle neutron scattering (SANS) technique, the Ca/Si ratio and density of C–S–H were reported as 1.7 and 2.6 g·cm–3 , respectively [60]. The morphology of C–S–H was also investigated at different Ca/Si ratios [61]. Two morphologies were distinguished, a branched structure for low Ca/Si ratio and ellipsoid particle structure at a high Ca/Si ratio. Masoumi et al. [62] investigated the variable chemical composition of C–S–H (with different Ca/Si ratio) and the chemical reaction of its nanolayer and aqueous solution. Kumar et al. [63] synthesized C–S–H with varying Ca/Si ratio from 1.0 to 2.0 and dynamic nuclear polarization (DNP) nuclear magnetic resonance (NMR) experiment was conducted to characterize the uniformity of the structure. In addition, the atomistic simulation along with DFT calculations were employed to test the structural stability of the proposed C–S–H. In MM simulations of hydrated cement phases, the ClayFF has proven to perform well in estimating its thermodynamic properties [64]. An empirical force field, named CSH-FF, which is a parametrized version of ClayFF, was employed to study the complex structure of C–S–H [65].
Tobermorite-like and jennite-like structures have been extensively used to simulate the cementitious C–S–H(I) and C–S–H(II) gels, respectively [66]. Different types of tobermorite minerals are known, including 9, 11, and 14 Å tobermorite (1 Å = 10–10 m), where the lengths in angstrom indicate the characteristic basal spacing [67,68]. Table 1 [37,69–71] presents the chemical structure and lattice parameters of mineral-like structures. The physical and chemical properties of used tobermorite minerals in simulations can be benchmarked to the experimental results reported in the literature [69]. Tobermorite with 14 and 11 Å structures were investigated using the CSH-FF model, and the results were compared with the DFT data [65]. Kova et al. [54] proposed three models that were based on the structure of tobermorite. These models of C–S–H were then revised by Kovacˇevic´ et al. [55], where periodic sets of models with different water contents were investigated. The ordered crystal (tobermorite models) and disordered glassy gels of C–S–H were investigated using the MD approach using the ReaxFF [72]. Another calcium silicate mineral, named jennite, was also utilized to simulate the structure of C–S–H [70].
《Table 1》
Table 1 Chemical structure and lattice parameters of minerals in hydrated cement.

3.2.2. Portlandite
CH, commonly referred to as portlandite, is the second most abundant hydration product, which accounts for about 15wt%– 25 wt% of the hydrated cement and has a hexagonal structure. It controls many durability aspects, including its role in buffering the pH, protecting the steel bars against corrosion due to the formation of a sub-micron thin protective Fe2O3 layer, reacts with siliceous materials (pozzolanic reaction) forming secondary C–S–H, affecting the pore structure, and so forth. However, when it is leached out the pore structure of cement may be less dense. It also acts as a buffer to damage C–S–H due to carbonation and acid and sulfate attack. Further, in the absence of portlandite, acids or sulfate ions will directly react with C–S–H, thereby destroying its binding capacity.
Due to the importance of CH in the durability of the cement matrix, the morphology of CH was examined using DFT and MD techniques through the use of ReaxFF [73]. It was reported that the Ca and O atoms on portlandite surfaces were attacked through nucleophilic and electrophilic forces. Such studies may help in better understanding the crystal growth of CH. With regards to the hydration of cement in the presence of nanoparticles, Tang et al. [74] studied the precipitation of CH from supersaturated solutions in the presence of sulfonated graphene nanosheets via the MD simulation. Additionally, the elastic properties of CH were predicted using DFT calculations [75].
The interaction between the growth of CH and silicate species, for example, C–S–H, would result in the comprehension of mechanisms and kinetics of cement hydration. Galmarini et al. [76] used the atomistic simulation technique to investigate the adsorption of silicate species on the surfaces of CH. The simulations provided useful information on the stability of Ca–Si complex and how it may influence the growth and hydration of Portland cement. Since the experimental assessment of the crystal growth of Portlandite in an aqueous system is difficult, the MD simulation approach can provide a fundamental understanding of the solid-solution interaction at the atomistic scale [77]. It was reported that under different adsorption sites, certain species control the growth of Portlandite, and in turn, this information will result in controlling the workability of cement. The diffusion of species in the nanopores of CH was investigated by Hou et al. [78]. Using the ClayFF, the MD study indicated that a strong layering of water on the surface of CH results in a high density and ordered organization. Consequently, the Cl– ions cannot stand for a long time on the surface of the weak H-bonds of portlandite.
《4. Mechanical and transport properties of cement paste》
4. Mechanical and transport properties of cement paste
《4.1. Structural and mechanical properties》
4.1. Structural and mechanical properties
The molecular simulation approach is a powerful technique that can be used to identify and quantify the structure and mechanical properties of cement-based materials. Few studies have been conducted in this direction. Nano-mechanical properties, including Young’s modulus (E), bulk modulus (K), shear modulus (G), and Poisson’s ratio of unhydrated and hydrated cements were evaluated. The elastic constants of the atomic structure are usually predicted by using the approach proposed by Theodorou and Suter [79]. Fig. 6 shows the elastic moduli (E, K, and G) of C–S–H crystals where the data were obtained from relevant literature [80]. Although there is quite scatter in the reported modeling results, the percentage of differences always remains less than 11%.
《Fig. 6》

Fig. 6. Elastic moduli of C–S–H crystals. Tob: tobermorite; the lines represent the fitted linear trends [80].
Generally, the presence of Ca atoms in C–S–H decreased the stiffness and cohesive force of the composite [81]. A higher Ca/Si ratio resulted in more structural defects in C–S–H [82]. In addition, it was reported that to improve the tensile strength of C–S–H, the lattice spacing between silicate chains should be smaller [83]. A study on the mechanical properties of C–S–H was conducted by Hou et al. [84]. The 11 Å tobermorite was used to simulate the C–S–H structure with different Ca/Si ratios (ranging from 1.3 to 2.0). The simulation results showed that the Ca/Si ratio has a significant effect on the stiffness and strength of C–S–H.
Hajilar and Shafei [68] used MD to predict the elastic properties of hydrated phases using analogue minerals, such as tobermorite, jennite, ettringite, kuzelite, and hydrogarnet. The simulation outcomes were compared with the experimental values in the literature; however, large differences were found. Consequently, to simulate the real structure of C–S–H, Hajilar and Shafei [68] tried to incorporate the gel porosity by employing the microporomechanics study. The obtained results were found to be in the range of values obtained by the nanoindentation technique. The micromechanics models were also used, in addition to the MD simulation approach, to obtain the mechanical properties of the two types of C–S–H, low density and high density [85]. The use of micromechanics technique is a general step to study a real amorphous structure of C–S–H from a multiscale view, which has not yet been fully understood. Recently, a combined simulation methodology that comprises the MD simulation for determination of elastic properties of cement paste and HYMOSTRUC3D method (a three-dimensional (3D) model that simulates the cement paste) for simulating the microstructure was proposed by Tavakoli et al. [86]. Besides, the lattice model was used to predict the stress– strain response of the cement paste. The obtained results were compared with the information available in the literature, and a good agreement was found.
Manzano et al. [87] tried to explain the discrepancy in the results of theoretical C–S–H and the actual values found experimentally. They reported that the elastic properties of 14 Å tobermorite and jennite were almost twice that of the C–S–H gel. Therefore, they stated that using the perfect crystalline structure of tobermorite and jennite is questionable. Also, they found that the composition of C–S–H, mainly the Ca/Si ratio and water/Ca (w/Ca) ratio, had a significant effect on the mechanical properties of the cementitious C–S–H gel. Although the inclusion of porosity in C–S–H (around 30% of the gel pores) was considered, the rescaled properties were still overestimated. Further, the defects and imperfections of the analogue minerals (tobermorite and jennite) were accounted for by a finite length of silicate chains. The study concluded that by introducing porosity along with crystalline defects, better comparable results could be obtained.
Additionally, the tensile strength and cohesion of hydrated cement are greatly affected by the structure and behavior of C– S–H at the fundamental scale [58]. A structure of C–S–H, in colloidal form, was investigated under uniaxial loading in different directions by Hou et al. [88]. The C–S–H was modeled using tobermorite. Since C–S–H has a layered structure, heterogeneous mechanical properties were obtained. It was reported that Young’s modulus in the Z-direction was 50% less than that in other directions and the authors related this to the presence of H-bonds and Ca2+ along Z-direction. In addition, the Ca–O bond has a significant influence on the tensile strength. The mechanical properties of the C–S–H structure with different Ca/Si ratios and subjected to tensile loading were also investigated by Hou et al. [89]. It was concluded that the high Ca/Si ratio degrades the tensile strength and elastic modulus of the gel. The crack growth mechanism under tensile loading of C–S–H with a central void, as shown in Fig. 7 [90], was investigated via MD simulation [90]. The simulation results showed that the presence of the centered nanopore decreased the stiffness and cohesive force of the C–S–H gel. Murray et al. [58] investigated the mechanical behavior of C–S–H subjected to the uniaxial tension and compression. They reported that the compressive and tensile strengths of C–S–H was an order of magnitude more than that of the cement paste at an engineering scale. The effect of the combination of tensile loading with water attack on the C–S–H gel was investigated using MD and grand canonical Monte Carlo (GCMC) approaches [91]. It was reported that the hydrolytic reaction due to water attack resulted in the loss of the cohesive stress in the tensioned gel. Zaoui [92] investigated the elastic behavior of 11 Å tobermorite crystal under the effect of external pressure with varying Ca/Si ratio. The results showed that the external pressure of 20 GPa is the threshold, and beyond this value, the elastic quantities changed. In addition, as the pressure was increased, the elastic properties of structures with Ca/Si ratio = 1.0 tended to be closer to those with a Ca/Si ratio of 0.83.
《Fig. 7》

Fig. 7. (a) C–S–H gel with a central void prepared for testing under uniaxial tension loading and (b) displacement patterns of Ca2+ under different strain rates at (i) 0, (ii) 0.16, (iii) 0.2, and (iv) 0.3 Å/Å. DSD: displacement standard deviation. Reproduced from Ref. [90] with permission.
The influence of temperature variation on the mechanical properties of C–S–H was also studied using MD simulation [93]. It was reported that the bulk and shear moduli of the composites decreased with an increase in the temperature. In a study by Honorio [94], the effects of both temperature and pressure on C–S–H were investigated using MC molecular simulations. It was reported that with an increase in temperature and pressure, the surface energy of the C–S–H decreased. The structure and properties of dry- and saturated-C–S–H gels were reported [95]. As observed in the macroscopic investigation and molecular simulation, the compressive strength of C–S–H in the saturated state is far more than its tensile strength.
While most of the MD simulation studies focused on the structural and mechanical properties of C–S–H, Lin et al. [96] reported on the dynamic mechanical behavior of C–S–H under shock compression loading. It was reported that the MD approach provides insight into the mechanisms of the shock wave and evaluates the related properties, such as the pressure, the specific volume, and the internal energy, which are difficult to measure experimentally.
Several force fields were used to evaluate the mechanical properties of cement compounds. Specifically, COMPASS, IFF, and universal force field (UFF) yielded better simulation results compared to the experimental outcomes [22]. The effects of the force field and supercell size are significant; consequently, these issues need to be addressed.
Fan and Yang [97] investigated the mechanical properties of C–S–H interfaced with water molecules and voids using the MD simulation. The ReaxFF was used in modeling C–S–H, and uniaxial tensile loading was applied along with different directions of the C–S–H structure. Five different atomic structures were modeled (Model I to Model V) that considered the varying lattice parameters and crystalline structures. It was reported that the fracture mechanics of C–S–H depends on the silicate chains while the strength is considerably decreased at the direction normal to silicate chains. Tavakoli et al. [82] used GCMC and MD approaches to study the effect of water on the elastic properties of C–S–H. In their study, the tobermorite and jennite structures were utilized to simulate the structure of C–S–H, and the COMPASS force field was employed. The results showed that increasing the w/Ca or Ca/Si ratios yielded in decreasing Young’s modulus. This is since at higher Ca/Si ratios, the structural defects of C–S–H become dominant, thereby decreasing the strength and Young’s modulus of the gel.
The stress–strain response of C3S, C2S, and C–S–H (tobermorite and jennite) were investigated using atomistic simulation [83]. The parameters, including strain rate and simulation box size, were also considered. The tensile stress–strain behavior of C–S–H (I) and C–S–H (II) were also investigated using ClayFF approach [98]. The deformation behavior of C–S–H, modeled as a jennite, under shear was studied by Rivos Murillo et al. [99]. It was reported that failure was initiated once the calcium oxide layers start slipping as the plastic deformation occurs. Such nanoscale findings may be used to prevent the layers from sliding and, thus, enhancing the macroscopic response of cement paste subjected to shear deformation. In a study by Manzano et al. [100], the shear strength of C–S–H gel under the influence of interlayer water was reported using non-equilibrium molecular dynamics (NEMD) approach. Such a study may be utilized in understanding the fundamental mechanisms of creep of the cement-based materials.
The constitutive behavior of C–S–H gel under combined loading, including compression or stretching and shear deformation, was investigated using molecular statics simulations [101]. It was reported that the pressure sensitivity of the shear strength and the heterogeneity on compressive and tensile strength were matched qualitatively with the nanoindentation results. The mechanical behavior of C–S–H, modeled as a jennite structure, was investigated under hydrostatic compression by Ref. [102]. It was reported that the constitutive relation of pressure and specific volume is almost linear, whereas a quadratic relationship was found in the specific internal energy and specific volume. An attempt was carried out by Espinosa et al. [103] to utilize the MD approach to propose a constitutive stiffness model of cement paste. To represent the matrix of cement paste at a certain degree of hydration, the proposed model was built on a binary composite system composed of C–S–H (jennite-like structure) and C3S or C2S. It was reported that the proposed composite system showed an isotropic behavior, although the individual phases have anisotropic characteristics. The authors related the isotropic response of the two-phase system to the interactions provided by the phases, and this behavior was linked to the macroscopic behavior of cement paste.
The fracture properties, such as surface energy, fracture toughness, and critical energy of C–S–H, are difficult to assess through experimental work. Consequently, MD simulation is a good technique to assess these properties [104]. It was reported that the fracture properties of cement materials at the atomic-scale would assist in the better design of structures with minimum materials. Nanoscale contact, including friction and scratch mechanisms, of C–S–H was studied using MD simulation by Jalilvand and Shahsavari [105]. Such deformation mechanisms on complex systems are challenging, therefore, the MD approach could be of great help in decoding the contact deformation mechanism.
The mechanical behavior of hydrated products of C3A, including hydrogarnet, ettringite, and monosulfoaluminate, was also studied via MD simulation [106]. The failure mechanisms of these important products under uniaxial tensile strains were described, and the stress–strain response was analyzed based on chemical bond and structural damage. Sarkar et al. [107] evaluated the deformation mechanism of ettringite as it is considered as one of the most important hydration products due to its role in the control of the rate of early hydration of cement. The IFF was used to study the molecular deformation of ettringite. The simulation process involves applying the uniaxial and triaxial tension and compression along all directions of the ettringite structure. It was reported that the adopted force field successfully simulated the elastic properties compared to the experimental data. In addition, the uniaxial tensile and compressive stress–strain curves confirmed the anisotropic property of the ettringite. The structural and mechanical properties of ettringite and thaumasite were also predicted using the DFT method. Based on DFT results, it was found that the thaumasite is stiffer than ettringite, and this was attributed to the low water content in the former.
《4.2. C–S–H–water interactions》
4.2. C–S–H–water interactions
Reactivity, dynamics, transport, and structural properties of interlayer water at the solid surfaces of cement were investigated. The mobility of water confined in C–S–H gel determines most of the chemical and physical properties (e.g., reactivity, shrinkage, and creep) of the binding phase (i.e., cement paste) [108]. The results obtained from MD simulation shed light on the position and mobility of H and O atoms of water molecules. The water confined in nano pores of C–S–H has hydrophilicity characteristics due to the availability of non-bridging oxygen atoms in the silicate chain, which serves as H-bonds [109]. In particular, since C–S–H has a highly reactive surface, a hydrolytic reaction takes place at the solid–liquid interfacial zone [110]. Therefore, the mobility of water molecules is restricted through the formation of H-bonds that are related to Ca–OH and Si–OH. In addition, it was found that upon increasing the Ca/Si ratio, the silicate skeleton in the C–S–H transforms from an ordered structure to an amorphous one. Among different atomistic water models to simulate the C–S–H structure, it was reported that the simplest water model, named single point charge (SPC), gives comparable results with good accuracy. Such information on the unique properties of water may be used in defining and explaining the change in the properties of hydrated cement.
The properties of water molecules are mainly influenced by the structure of the interesting substrate. It was reported that the water molecules confined in C–S–H gel dissociate into hydroxyl ions and thereby attack the Si–O and Ca–O bonds [111]. In this regard, Manzano et al. [109] studied the mechanism of water dissociation in the C–S–H gel using the MD approach through the ReaxFF. It was reported that upon dissociation of C–S–H, the formation of Ca–OH and Si–OH groups did not influence the mechanical properties while the shear strength was greatly affected. The effect of elevated temperature on the interlayer water dissociation of C–S–H was also investigated [112]. The water confined in C–S–H nanopores is temperature-dependent, where its mobility increased with temperature. The MD simulation provides insights into the breakage of silicate chains at an elevated temperature where the hydrolytic reaction of water decreases the ionic–covalent bonds in the C–S–H. However, the effect of pressure on water dissociation in hydrated cement needs to be investigated.
Yoon and Monteiro [113], in their study on water confined in the layers of 14 Å tobermorite, found that upon increasing the temperature from 100 to 300 K, water mobility increased while its structure kept unchanging. The hydrolytic weakening of water on quartz minerals under uniaxial tension was investigated [114]. The GCMC approach was used to simulate the water adsorption and the dynamics, structure, and mechanical properties of the system. Tang et al. [115] investigated the transport properties of water molecules through the C–S–H surfaces under tensile loading by employing ClayFF. A C–S–H model with a nanopore of 40 Å was constructed, as shown in Fig. 8 [115]. The authors stated that the selected force field provides good insight into the interaction between the substrate and liquid phases; however, they recommended using the ReaxFF to further investigate the reactivity of the system.
《Fig. 8》
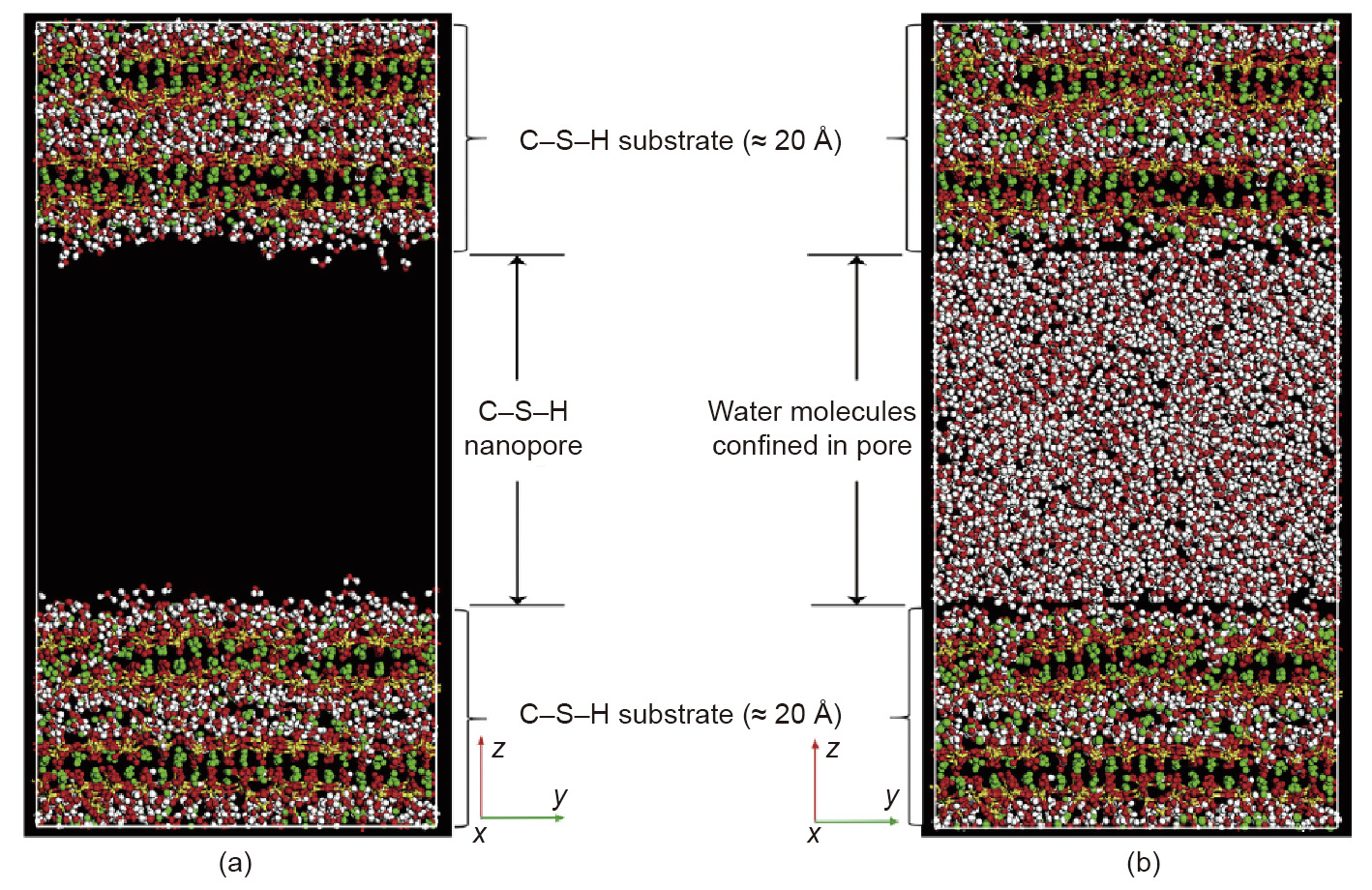
Fig. 8. (a) Nanopore C–S–H model and (b) water molecules confined in the pores Reproduced from Ref. [115] with permission.
《4.3. Transport mechanisms of C–S–H》
4.3. Transport mechanisms of C–S–H
The durability of hydrated cement is dependent on the transport mechanisms and interfacial adsorption of water molecules and chemical species [116]. The atomic-level simulation of the interaction between cement phases and other species has resulted in a much better understanding of the long-term durability of concrete. The migration of water molecules and ions through a porous cementitious medium was numerically studied through molecular simulation. Specifically, the MD simulation approach provided different molecular mechanisms of water and ionic diffusion through the C–S–H gel pore [117]. The dynamics and structure of water and ions in the pore solution of cement paste were studied using MD simulations [118]. Generally, the mean squared displacement (MSD) technique is used to obtain self-diffusion coefficients. Moreover, the evolution of the ionic conductivity on the pore solution is also discussed [118].
In some studies, the simulation results were compared with the available experimental data. Hou and Li [119] utilized the jennite crystal structure of the C–S–H and examined the transport of water and other ions in its nanopores. In their model, the MD technique was used based on ClayFF. The authors claimed that the proposed model matches well with the results of 35Cl NMR studies. In another study [120], the authors showed that there was a significant reduction in the diffusion of chloride ions. Further, it was reported that the results of simulation studies match well with the experimental data obtained by NMR and quasi-elastic neutron scattering (QENS) techniques. They also reported that with increasing distance from the channel, the structural and dynamic behavior of the water molecules varies and gradually translate into bulk water properties at distances of 10–15 Å from the liquid–solid interface.
The transport mechanisms of chloride and sulfate ions in the interlayers of C–S–H were also investigated by Hou et al. [121]. The C–S–H models with nanopore channels were first constructed, and then the capillary flow of water and ions was simulated, as shown in Fig. 9 [121]. The results indicated that the sulfate ions migrate slowly than the chloride ions. The chemical attack was simulated through the MD simulation and experimentally investigated by active acoustic monitoring. It was reported that the ionic–covalent bonds in the C–S–H gel were greatly affected by the sulfate attack. The transport of chloride ions and water molecules through C–S–H gel (modeled as 11 Å tobermorite) was studied by Zhou et al. [122] by employing ClayFF. The authors reported that the presence of calcium ions improved the chloride binding capacity of cement. The influence of ions, such as Ca, Mg, Al, and Na, confined in the inlayers of C–S–H, on the cohesive strength was also studied by Hou et al. [123]. Additionally, the capillary adsorption of sodium chloride solution in the nanopores of C–S–H gel was discussed by Hou et al. [124]. In a study by Yang et al. [125], the effect of the size of C–S–H gel nanopores on the transport properties of NaCl solution was investigated. Different C–S–H models, considering pore size ranging from 1 to 3.5 nm, were adopted. It was concluded that the size of the gel pores decreased as they were blocked by the mobility of chloride and sodium ions.
《Fig. 9》

Fig. 9. (a) Unsaturated transport model (C–S–H gel pore with the bottom solution) and (b) capillary adsorption process of NaCl + Na2SO4 solution through the pore at different times. Reproduced from Ref. [121] with permission.
Yang et al. [126] performed an MD study on the adsorption behavior of sulfate and sodium ions in the nanometer channel of C–S–H gel under different Ca/Si ratios. They used a saturated nanopore model to simulate the sulfate and sodium ions solvated in the nanometer channels of C–S–H, as displayed in Fig. 10 [126]. The results pointed towards two adsorption mechanisms: SO42– ions interacted with the calcium ions while Na+ ions linked to the non-bridging oxygen sites. Zhou et al. [127] also examined the effect of the Ca/Si ratio on the transport behavior of water molecules and ions (calcium and chloride). It was observed that with increasing Ca/Si ratio, the long silicate chains were broken to provide more oxygen sites to adapt the water and calcium ions and thereby yielded more adsorption of Cl–. The alkali adsorption through the surface of C–S–H was explored via semi-grand canonical MC and MD simulations and mesoscale modeling [128]. It was realized that the adsorption of sodium and potassium increased as the Ca/Si decreased.
《Fig. 10》

Fig. 10. C–S–H saturated nanopore model with confined water molecules and sodium and sulfate ions. Reproduced from Ref. [126] with permission.
The diffusion of sodium chloride solution under elevated temperature through C–S–H gel was investigated by Ref. [129]. The COMPASS force field was adopted, and the C–S–H structure was modeled based on tobermorite and jennite minerals. The results indicated that the Na+ ions demand more activation energy to diffuse than the Cl– ions. The diffusion coefficients and activation energies of ions were obtained and compared with the available experimental data. The transport behavior of NaCl solution through the nanopores of C–S–H under an external electric field ranging from 0 to 0.05 V·Å–1 was also investigated [130]. Such research can provide useful information on the diffusion of ions in cement under electrochemical desalination processes.
In addition to the durability of C–S–H, other cement phases were also reported. The chloride ion binding on the surfaces of hydrated cement paste, including tobermorite, portlandite, and ettringite, was investigated using MD through ClayFF [131]. It was demonstrated that the Cl– binding capacity was in the following decreasing order: portlandite, ettringite, and tobermorite. This finding is in good agreement with the experimental outcomes from 35Cl NMR. The transport of water molecules and sodium and chloride ions through the monosulfoaluminate (AFm phase) was undertaken by Hajilar and Shafei [132]. The authors concluded that the inner- and outer-sphere formations govern the diffusion of sodium and chloride ions where mobility of Cl– is faster than that of Na+ . Huang et al. [133] reported on the doping of iron in aluminate phases, mainly of hydrogarnet [C3(A,F)H6], and its role in the sulfate attack. The simulation results based on COMPASS force field revealed that the iron-containing phases possessed better sulfateresistance than the iron-free phases. The adsorption and transport mechanism of ions on the nano pores of Friedel’s salt (product formed due to the reaction of C3A with chloride ions), were also studied using MD simulation [116]. It was observed that the immobilizing ability of ions on the surface of Friedel’s salt is strong whereas the dynamics capability of water molecules is larger than ions.
The interaction between the cement-based materials and other materials was also modeled using atomistic simulation. Work on the utilization of cementitious materials (C–S–H and 9 Å tobermorite) to encapsulate the radioactive strontium-90 was reported by Ref. [134]. Additionally, Bu et al. [135] examined the adsorption behavior of cesium ions, a major by-product of nuclear fission, in C–S–H (tobermorite and jennite). It was elucidated that tobermorite strongly adsorbed cesium than jennite. Jiang et al. [136] used MD simulation based on COMPASS force field to explore the interaction between C–S–H and lauric acid (C–S–H@LA). Such a composite could be utilized in thermal energy storage (TES), which is one application of phase change materials (PCMs).
《5. Atomistic simulation on low-carbon binders》
5. Atomistic simulation on low-carbon binders
Nowadays, supplementary cementitious materials (SCMs) are commonly used to reduce the environmental impact of cement production [137]. SCMs, such as fly ash and slag, have active aluminate minerals which are incorporated into C–S–H to produce calcium alumino–silicate hydrate (C–A–S–H) gel. In alkali-activated binders, including geopolymers, the activation of fly ash with NaOH produced sodium aluminosilicate hydrate gel (N–A–S–H).
The molecular simulation has been applied to investigate the geopolymers, which have physical and chemical similarities to the hardened Portland cement. The lack of linking the chemical compositions of such materials and macroscopic performance limited their applications in the construction industry. Thus, molecular simulation could be an effective way to better understand the nano-scale mechanisms and important structural features of these binders [138]. The effects of water content and Si/Al variation on the mechanical properties of geopolymer binder were investigated through MD simulation by Sadat et al. [139]. The pair distribution function (PDF) was used to validate the simulation model. It was reported that increasing the Si/Al ratio increased the stiffness and tensile strength of the binder. Water and chloride ions diffuse in nanopores of geopolymer gel (A–S–H) using COMPASS force field [140]. In the simulation system, the geopolymer gel (A–S–H) was assumed to be in the form of C–S–H layers, and water molecules and chloride ions had been implanted into the initial structure of zeolite (a mineral analogy of geopolymer gel). Based on the thermodynamics analysis, it was reported that the geopolymer gel was less likely to absorb chloride ions as the temperature increased, therefore, corrosion tends to occur in other parts of a structure.
Lyngdoh et al. [141] proposed a layered but disordered structure of N–A–S–H gel where water molecules are confined in the interlayer. The transportation of ions through the N–A–S–H gel was demonstrated through the MD approach [142]. Due to the presence of non-bridging oxygen sites in the N–A–S–H structure, strong H-bonds were formed. The interaction of water and sulfate and magnesium ions with the N–A–S–H gel was also investigated by employing ClayFF. Additionally, the structure of N–A–S–H gel exposed to elevated temperature was studied utilizing the ReaxFF [143]. It was concluded that at a higher temperature (ranging from 300 to 1500 K), the water molecules decompose, and both Al–O–Si and Si–O–Si bonds are broken, thereby transforming the gel to a weak chain structure. In addition, aluminosilicate skeleton under elevated temperature and subjected to tensile stresses become weak due to hydrolytic reactions that deteriorate the gel structure. The degradation of tensile behavior of N–A–S–H gel due to hydrolytic reaction was studied using MD simulation by Zhang et al. [144]. It was observed that the confined water is physically and chemically crosslinked to the Si–Al framework.
The use of atomistic simulation techniques to study the C–A–S–H gel, a product of alkali-activated slag (AAS), was also reported. C–A–S–H gel has longer chains than the C–S–H gel, where the silicon in bridging positions of the latter is replaced by alumina [145], as illustrated in Fig. 11 [146]. ReaxFF was used to study the polymerization and hydrolytic reactions of C–A–S–H gel. The presence of Al atoms in the interlayer of C–A–S–H gel enhances the structure and mechanical properties of the binder, that is, C–A–S–H. The C– A–S–H gel was observed to be stable at higher Al/Ca ratios. In addition, with increasing Al content, Young’s modulus and tensile strength improved. In a study by Yang et al. [147], C–A–S–H gel was modeled with different Al/Si ratios ranging from 0 to 0.2. It was reported that the incorporation of aluminate species yielded more crystalline order with improved connectivity of the Q species (the connectivity factor). Further, at a higher Al/Si ratio, more dissociation of interlayer water molecules occurred as a result of increased reactivity of the bridging oxygen sites in Si–O–Al. Also, Wan et al. [148] concluded that the availability of Al and Si sites in C–A–S–H gel increased the mean silicate chain length of the structure. Therefore, the authors suggested that the mean chain length could be used as a criterion to improve the performance of alkali-activated binders. Zhang et al. [149] conducted a study on the effect of temperature on the C–A–S–H gel. It was concluded that the H-bonds are destroyed at elevated temperatures and thereby resulting in the observed expansion.
《Fig. 11》
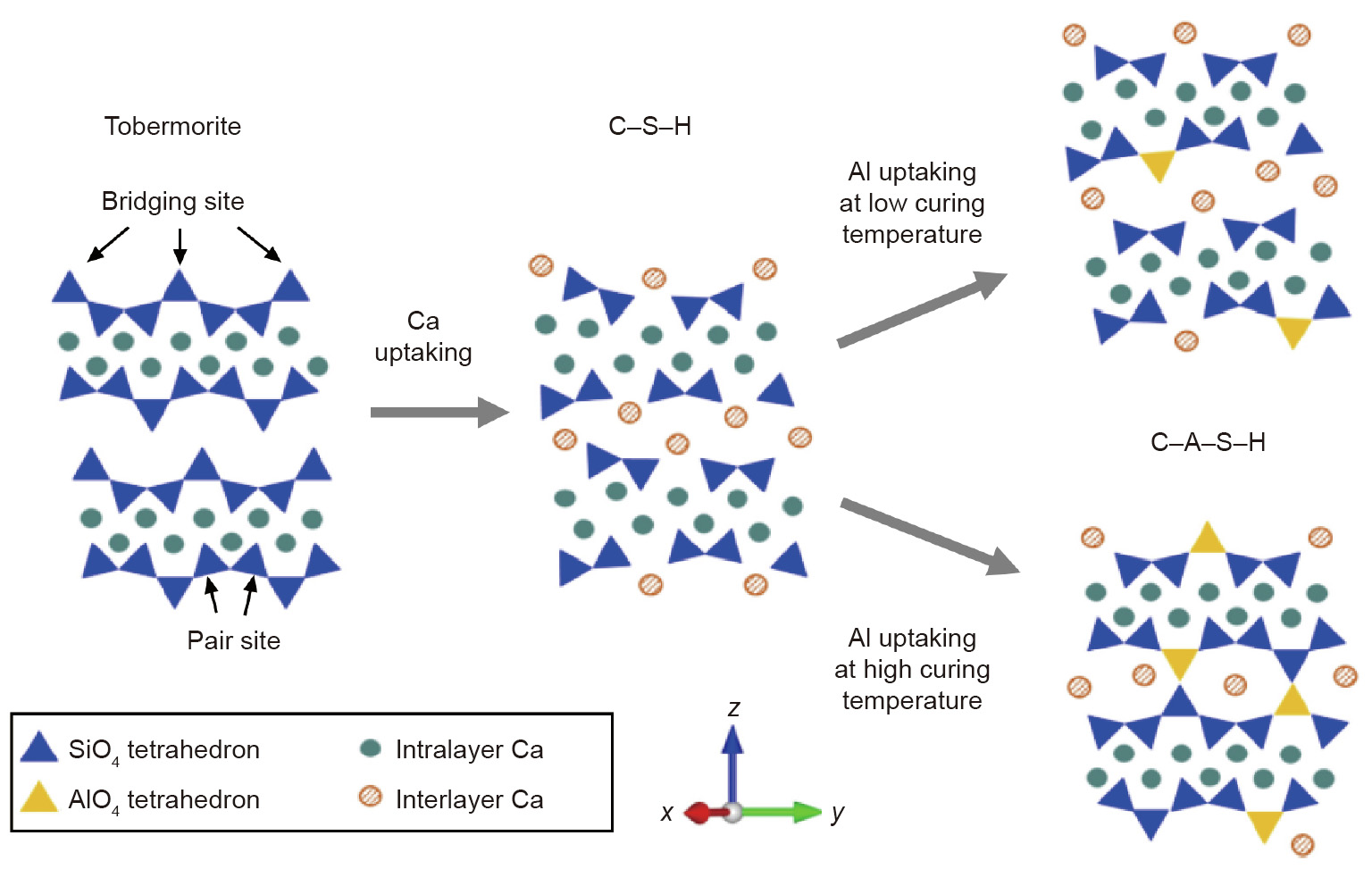
Fig. 11. C–A–S–H structure after uptaking of Al. Reproduced from Ref. [146] with permission.
The transport mechanism of NaCl solution through the C–A–S–H gel was studied using the MD method [150]. Tobermorite was adopted to model C–S–H, and then C–A–S–H was constructed by replacing silicon on the surface with aluminum. The simulation was performed through ClayFF with a constant number of atoms, constant volume, and constant temperature (NVT) ensemble. The results confirmed that, using nano-scale investigation, the presence of Al ions in the C–A–S–H structure had restricted the ingress of water molecules and sodium ions through the nanopores of C–A–S–H. In addition, the Al–Si substitution provides sites for forming oxygen leading to Na–O stable bond. In another study, the effect of NaCl solution with aluminates on the properties of nanopores of C–S–H and C–A–S–H was investigated by Hou and Li [151]. It was reported that, in C–A–S–H, the confined layered water is densified, and this is since aluminate silicate chains having more H-bonds were contributed by oxygen atoms. Further, compared to C–S–H, C–A–S–H immobilized more sodium and chloride ions.
Ding et al. [152] investigated the mechanisms of sulfate attack in Portland cement paste admixed with granulated blast furnace slag (PC–GBFS). The molecular structure of C–S–H was constructed in analogue to 11 Å tobermorite, while C–A–S–H was structured with more bridging aluminate tetrahedron sites. The sodium and sulfate ions (Na2SO4) were added to the pores to simulate the sulfate transport mechanism. ClayFF was utilized in this simulation. The MD simulation results shed light on the decalcification mechanism in C–A–S–H due to sulfate ions, while the addition of GBFS stabilized the alumino–silicate chains. In another study, the interaction of sulfate ions with C–A–S–H gel was also investigated through the MC method combined with MD simulation [153]. The structure of C–A–S–H was built using the crystalline C–S–H gel, where the water molecules and sodium sulfate (5 wt% Na2SO4) were added. The atomistic simulations provided some molecular insights on the mechanisms of sulfate attack on C–A–S–H gel. Additionally, the MD simulations on other binders were also reported. Magnesium phosphate cement (MPC), as an example, was simulated through its main hydration product (i.e., struviteK crystal) to explore its nano-scale mechanisms and mechanical behavior. The properties of pozzolanic concrete were also investigated via MD simulation. Specifically, the pozzolanic activity indices of different pozzolanic materials were assessed based on the water–surface interaction energy theory. Therefore, based on the simulation study, a new equation was proposed for obtaining the unique activity index, which would simplify the selection of desired pozzolanic materials. Therefore, nano-scale simulations may offer accurate prediction of pozzolanic activity of pozzolanic materials [154].
《6. Concluding remarks》
6. Concluding remarks
The application of nanotechnology in cement and concrete science is still in a preliminary stage. Research in nanoengineering through molecular simulations and modeling of cement-based materials has gained attention in recent years. This paper highlights the importance of molecular investigations of the complicated cement system at the fundamental scale and it extensively reviews the studies that have been carried out in this area. While most of the studies focused on the mechanical and transport properties of C–S–H gel, its composition and structure have not yet been known. Therefore, the research in computational modeling of cement-based materials is still in progress, and more efforts need to be orchestrated in developing realistic atomistic models of hydrated cement phases, mainly C–S–H. So far, the molecular information obtained from the atomistic simulations is useful in explaining the microscopic properties that control concrete behavior at the engineering scale. Further, the durability characteristics evaluated by the MD simulation have provided useful insights on the nanoscale mechanisms of transport and diffusion of water and ions through the cementitious materials. In summary, research and development in computational materials science of cement and concrete (CMS-CC) should be encouraged to assist the optimization of experimental work. Needless to say, the research on nano-scale simulation would ultimately enable researchers and engineers to design new cement mixtures or produce a new generation of construction materials that can suit the requirements of workability, strength, durability, and volume stability for sustainable construction that will lead to economic, technical and environmental benefits.
《Acknowledgment》
Acknowledgment
The authors would like to acknowledge the support provided by the Deanship of Research Oversight and Coordination (DROC) at King Fahd University of Petroleum and Minerals (KFUPM), Saudi Arabia, for funding this work through Project No. DF191009. The support provided by the Department of Civil and Environmental Engineering and Interdisciplinary Research Center for Construction and Building Materials at the Research Institute, KFUPM, is also acknowledged.
《Compliance with ethics guidelines》
Compliance with ethics guidelines
Ashraf A. Bahraq, Mohammed A. Al-Osta, Omar S. Baghabr, Al-Amoudi, I. B. Obot, Mohammed Maslehuddin, Habib-ur-Rehman Ahmed, and Tawfik A. Saleh declare that they have no conflict of interest or financial conflicts to disclose.
《Appendix A. Supplementary data》
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.eng.2021.06.023.











 京公网安备 11010502051620号
京公网安备 11010502051620号




