

《1. Introduction》
1. Introduction
Chronic hepatitis caused by hepatitis C virus (HCV) infection is a serious global issue, since chronic hepatitis C is the main cause of both liver cirrhosis and hepatocellular carcinoma (HCC). According to a report from the World Health Organization (WHO) in 2017, 71 million people worldwide (1%) are estimated to be infected with HCV [1]. Approximately 30% of HCV-infected individuals display persistent chronic hepatitis, which may progress to liver cirrhosis and HCC [2].
HCV is a member of the genus Hepacivirus within the Flaviviridae family, which is a group of small, enveloped, single-stranded RNA viruses [3]. HCV particles are formed from structural proteins, including the HCV core protein and the envelope glycoproteins E1 and E2. Since nonstructural proteins (p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B) play an important role in the HCV life cycle, they have become molecular targets for direct-acting antiviral agents. For example, NS4B regulates replication complex formation, NS5A is the main component of HCV RNA replication and particle production, and NS3–NS5B are responsible for the replication module and catalyze the amplification of the viral RNA genome [3,4]. HCV contains a 9.6 kilobase (kb) positive-strand RNA genome composed of a 5' noncoding region (NCR) with an internal ribosome entry site, an open-reading frame that encodes the structural and nonstructural proteins, and a 3' NCR [5] (Fig. 1).
《Fig. 1》

Fig. 1. Structure of the HCV and its core protein. The HCV genome (structural genes and nonstructural genes) encodes a polyprotein of 9.6 kb flanked at both ends by 5' and 3' untranslated regions (UTRs), This polyprotein is processed by cellular and viral proteases into ten structural and nonstructural proteins (Core, E1, E2, p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B). The core protein is released from a polyprotein by a signal peptidase (SP). The precursor core of 191 aa is processed by a signal peptide peptidase (SPP) to obtain a mature protein of approximately 177 aa, which is composed of two domains (D1 and D2). The D1 domain is divided into basic domain 1 (BD1), basic domain 2 (BD2), and basic domain 3 (BD3). The D2 domain includes helix I (HI) and helix II (HII), which are separated by a hydrophobic loop (HL). Reproduced from Ref. [4] with permission of Springer Science Business Media New York, © 2014 and from Ref. [5] with permission of Elsevier, © 2011.
Previous studies have demonstrated a strong association between HCV infection and various metabolic diseases, including hepatic steatosis, type 2 diabetes mellitus (T2DM), and iron and porphyrin accumulation [6,7]. However, metabolic disorders are also strongly influenced by daily lifestyle, such as overeating, high-fat and high-carbohydrate diets, and lack of exercise. Moreover, persistent HCV infection is often accompanied by hepatic inflammation and fibrosis, disrupting insulin signaling and lipid metabolism through inflammatory cascades and hepatocyte injury [8–12]. It is therefore difficult to understand the singular direct contribution of HCV to metabolic diseases. In order to decipher the pathogenic principles of HCV, a variety of HCV transgenic mice models have been developed using the ten structural and nonstructural proteins [13–15]. The fact that only HCV core protein transgenic mice exhibited spontaneous glucose intolerance, hepatic steatosis, and HCC similar to those seen in HCV-infected patients [16] corroborated the crucial role of the HCV core protein in HCV-related metabolic disturbances.
In this review, we summarize the recent key findings obtained from mouse models transgenic for the HCV genome, with a special focus on characteristic observations in HCV core gene transgenic mice, and discuss the mechanisms of steatosis-derived hepatocarcinogenesis induced by the HCV core protein. For an objective and comprehensive review, we entered several keywords on this theme (HCV transgenic mice, fatty liver, HCC, fatty acid (FA), cholesterol, calorie restriction, dietary intervention, etc.) into PubMed and searched for related articles published between 1995—when HCV protein transgenic mice were first established [14]—and 2021.
《2. Function of the HCV core protein》
2. Function of the HCV core protein
《2.1. Structure of the HCV core protein》
2.1. Structure of the HCV core protein
The HCV core protein is a 21-kilodalton (kDa) multifunctional protein with lipid and RNA binding activity, whose main function is the construction of a viral capsid to cover and protect genomic RNA while simultaneously transmitting the virus from one cell to another [4,17]. After the precursor of the HCV core protein (191 amino acids (aa)) is released from a polyprotein, further processing by a signal peptide peptidase produces the mature core protein form [4].
The HCV core protein consists of two domains: hydrophilic D1 and hydrophobic D2. The D1 domain is composed of three basic clusters: basic domain 1 (BD1; 2–23 aa), basic domain 2 (BD2; 38–74 aa), and basic domain 3 (BD3; 101–121 aa). The D1 domain is related to the oligomerization necessary for particle formation and RNA binding [4]. The D2 domain is responsible for associations with the endoplasmic reticulum (ER) and lipid droplets (LDs). The main structural element in D2 consists of two amphiphilic α helices (helix I (HI) and helix II (HII)) separated by a hydrophobic ring. The two helices can fold in a hydrophobic environment, thus implicating the interaction of lipids in maintaining structural integrity. Mutational studies have shown that the combination of HI, the hydrophobic loop, and HII is inseparable from the formation of LDs [18] (Fig. 1). The approximately 20 final aa of the hydrophobic D2 domain serve as the signal sequence targeting the envelope glycoprotein E1 [4].
《2.2. Role of the HCV core protein in cells》
2.2. Role of the HCV core protein in cells
To evaluate the direct function of the HCV core protein in cells, in vitro experiments using cultured cells stably overexpressing the HCV core protein are indispensable. In the QSG7701 humanderived non-tumor liver cell line, expression of the HCV core protein inhibited cell apoptosis [19] by disrupting the retinoblastoma tumor suppressor protein (pRb)/ E2F transcription factor 1 (E2F-1) balance [20]. In contrast, the translocation of B cell leukemia/lymphoma 2-associated X protein (BAX) from the cytoplasm to the mitochondria, destruction of the mitochondrial membrane potential, release of cytochrome c, and activation of caspase-9 and caspase-3 by the HCV core protein appeared to drive apoptosis [21]. The HCV core protein not only upregulated nuclear factor (NF)-jB to suppress host cell responses, but also enhanced autophagy by increasing Beclin-1 expression [19]. Although autophagy can help eliminate pathogens, HCV can accelerate self-replication through autophagy, leading to HCV survival and persistent infection [19].
In HCC cell lines, the HCV core protein significantly enhanced Wnt/β-catenin signal transduction activity, a key driver of hepatocyte proliferation and hepatocarcinogenesis [22]. It also regulated Wnt1 in HepG2 cells to promote abnormal proliferation [23]. Several studies have shown that the HCV core protein can enhance the expression of activator protein (AP)-1 and vascular endothelial growth factor (VEGF) in liver cancer cells, indicating that HCVinduced angiogenesis in HCC might be partially mediated by this protein [24]. The HCV core protein has the ability to upregulate hypoxia-inducible factor-1α under hypoxic conditions, thereby helping to increase VEGF expression [25]. A report has also shown that the HCV core protein promoted the proliferation of human liver cancer cells by activating NF-κB and upregulating the expression of tumor necrosis factor (TNF)-α. The HCV core protein stimulated hepatocyte proliferation and chemoresistance by inhibiting nuclear receptor subfamily 4 group A member 1 (NR4A1) [26]. Hepatoma cells expressing the HCV core protein activated cocultured stellate cells in a manner mediated by transforming growth factor β [27]. The interaction between the HCV core protein and LDs plays an important role in the process of HCV infection. The HCV core protein has been shown to affect sphingolipid and cholesteryl ester biosynthesis, which was partially associated with abnormal lipid metabolism in HCV-infected patients [28]. In addition, the expression of insulin-like growth factor-binding protein (IGFBP)-1 was significantly reduced in hepatocytes transfected with the HCV core protein [29]. Since IGFBP-1 may be a key factor in hepatic insulin sensitivity and glucose metabolism [30], this effect may contribute to insulin resistance in chronic hepatitis C.
《3. Generation of HCV core gene transgenic mice》
3. Generation of HCV core gene transgenic mice
《3.1. Why are HCV core gene transgenic mice necessary?》
3.1. Why are HCV core gene transgenic mice necessary?
Since the results of cell experiments overexpressing HCV protein are obtained under specific conditions, in vivo animal experiments are extremely valuable in evaluating the impact of HCV protein in the whole body by mimicking persistent HCV infection in humans. However, HCV cannot infect rodents, the conventional animals of most in vivo experiments [31]. As an alternative, a variety of HCV protein transgenic mice have been generated for understanding the pathogenesis of HCV proteins in vivo [13,14,32].
The establishment of HCV mouse models has helped to reproduce the clinical features of HCV-infected patients and assess the pathogenesis of chronic HCV infection. Transgenic mice carrying the HCV envelope genes E1 and E2 were first documented in 1995 [14]. Immunostaining showed that the envelope protein was localized mainly in the cytoplasm of hepatocytes around the hepatic central veins. There was no detectable evidence of liver disease until 16 months of age, although the animals exhibited exocrinopathy involving the salivary and lachrymal glands, indicating the direct involvement of HCV in the pathogenesis of sialadenitis in HCV-infected humans [33]. The HCV NS5A protein has numerous hallmark characteristics, including the sequestration of p53 in the cytoplasm, downregulation of the p21 protein, activation of signal transducer and activator of transcription 3 (STAT3), and inhibition of TNF-α-mediated apoptosis. However, HCV NS5A protein gene transgenic mice did not show obvious pathogenicity [13], which was also the case for HCV NS3A/NS4Aexpressing transgenic mice [32,34].
Among the HCV structural and nonstructural proteins, mutations are rare in the core gene, suggesting that the core protein play a key role in the pathogenesis of HCV [35,36]. The ability of the HCV core protein to regulate a variety of signaling pathways in the host, such as those related to apoptosis, gene transcription, cell transformation, and immune responses [37–39], is also related to hepatocarcinogenesis [16]. Therefore, clarifying the role of the HCV core protein in vivo using transgenic mouse systems such as the one established in 1997 will provide clues about HCV pathogenesis [40].
《3.2. How were HCV core gene transgenic mice generated?》
3.2. How were HCV core gene transgenic mice generated?
To generate HCV core gene transgenic mice, the expression vector pBEPBglII containing hepatitis B virus regulatory elements was used in the plasmid construction process [14]. By double digestion with PstI and EcoRI, a (1 ± 6) kb fragment containing the core protein coding region was excised from plasmid pSR39 [41] after treatment with T4 DNA polymerase and the attachment of BclI linkers. Subsequently, it was connected to the BglII site of plasmid pBEPBglII. A (1 ± 2) kb KpnI–HindIII fragment from pBEP39 was purified by means of polyacrylamide gel electrophoresis (PAGE) and microinjected into mouse embryos from C57BL/6N mice. Then, 1 ng of tail DNA was amplified by polymerase chain reaction (PCR) to identify transgenic mice [40] (Fig. 2).
In the strain of HCV core gene transgenic mice developed by Moriya et al. [40], core protein expression in the liver begins at birth. This transgenic mouse line has levels of core protein similar to those of HCV-infected humans. The mice begin to exhibit LDs in liver cells 3 months after birth, and liver steatosis continues as the animals grow. Hepatic adenomas containing fat droplets in the cytoplasm appear at approximately 16 months of age, and the mice eventually exhibit HCC at the age of 17 months [16].
《Fig. 2》
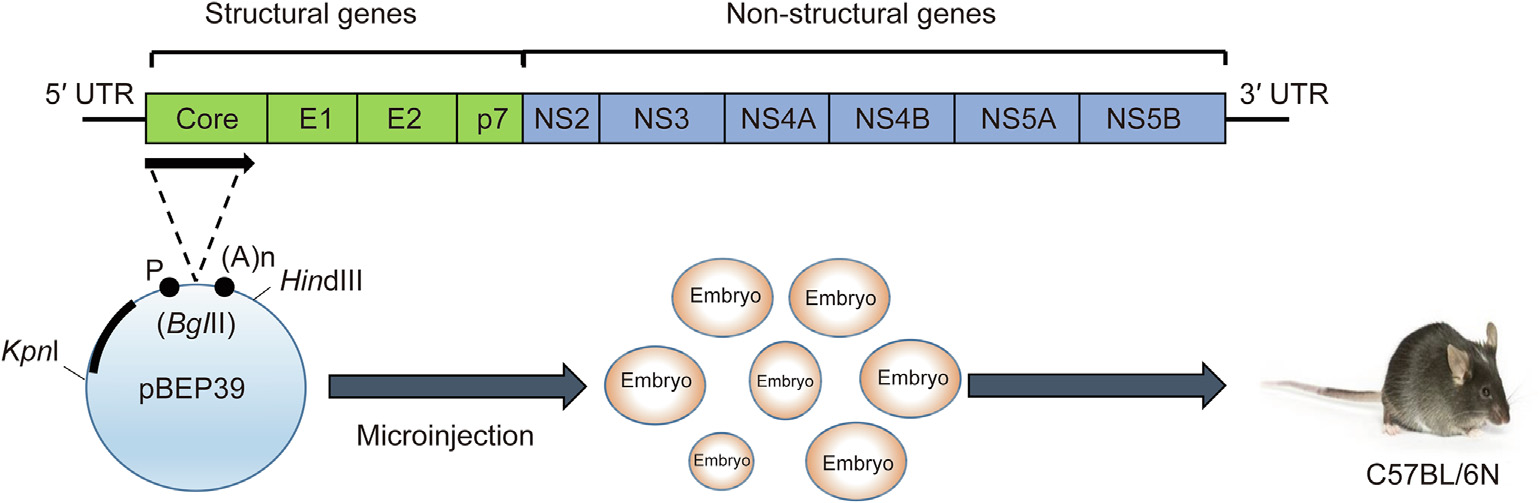
Fig. 2. Generation of HCV core gene transgenic mice. pBEP39 is an expression vector containing hepatitis B virus regulatory elements. A (1 ± 2) kb KpnI–HindIII fragment is microinjected into mice embryos, which are then introduced into C57BL/6N mice. P: promoter; (A)n: polyadenylation signal. Reproduced from Ref. [5] with permission of Elsevier, © 2011 and from Ref. [40] with permission of Society for General Microbiology, © 1997.
《3.3. Hepatic insulin resistance in HCV core gene transgenic mice》
3.3. Hepatic insulin resistance in HCV core gene transgenic mice
T2DM is a complex multi-system disease involving defects in insulin secretion, which increases hepatic glucose production and insulin resistance [42,43]. Although a positive association between T2DM and chronic hepatitis C has been reported in epidemiological studies, it remains unclear how HCV contributes to the onset of T2DM. A significant and direct mechanistic link between insulin signaling and HCV was first demonstrated using HCV core gene transgenic mice [40]. Whereas the body weight of the transgenic mice was similar to that of control mice, their serum insulin levels and pancreatic β cell mass were both significantly higher. Hyperin sulinemia was observed as early as at 1 month of age in the transgenic mice and preceded hepatic steatosis, demonstrating the direct and initial action of the HCV core protein in insulin resistance. TNF-α is a key contributor to insulin resistance. In fact, blocking TNF-α action restored hepatic insulin sensitivity in the transgenic mice, confirming that disrupted hepatic insulin signaling by the HCV core protein was mediated by TNF-α [44]. The HCV core protein may also stimulate the activation of proteasome activator 28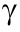 (PA28) to affect insulin signal transduction [45]. Although the mechanism of insulin resistance is multifactorial [42,43,46,47], the above findings support the direct action of the HCV core protein on hepatic insulin resistance and T2DM development, which are frequently accompanied by chronic hepatitis C [12] (Fig. 3).
(PA28) to affect insulin signal transduction [45]. Although the mechanism of insulin resistance is multifactorial [42,43,46,47], the above findings support the direct action of the HCV core protein on hepatic insulin resistance and T2DM development, which are frequently accompanied by chronic hepatitis C [12] (Fig. 3).
《Fig. 3》
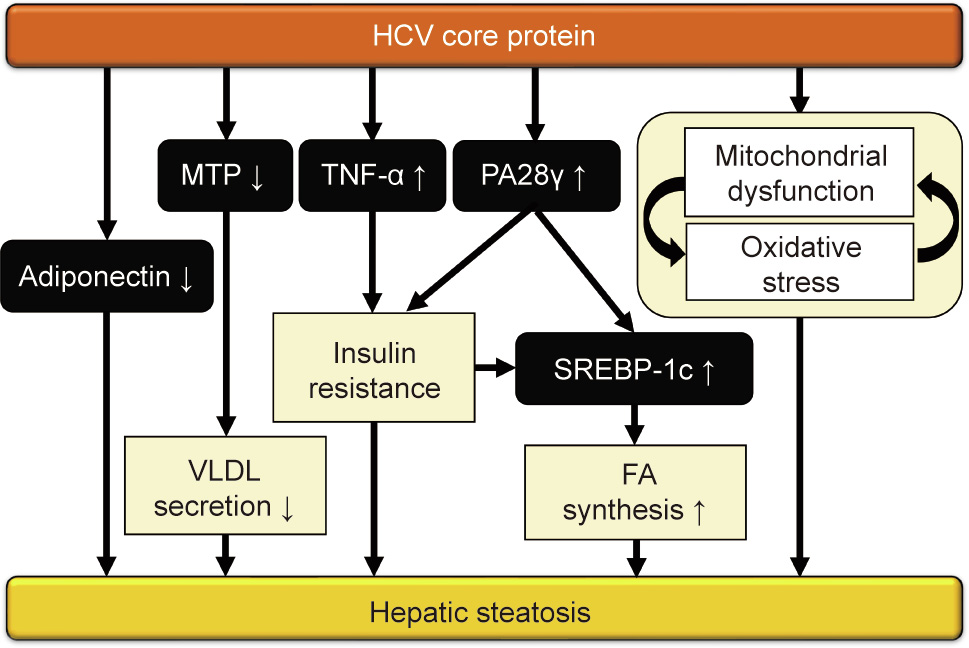
Fig. 3. Mechanism of hepatic steatosis induced by the HCV core protein. The HCV core protein induces hepatic steatosis in transgenic mice via several pathways. TNF-α and PA28 cause insulin resistance, upregulate sterol-response elementbinding protein (SREBP)-1c, and stimulate lipogenesis. Decreased adiponectin and microsomal triglyceride transfer protein (MTP) expression also leads to diminished very-low-density lipoprotein (VLDL) secretion and hepatic steatosis. Mitochondrial dysfunction by the HCV core protein can reduce FA catabolism and enhance oxidative stress, thereby aggravating hepatic steatosis.
《3.4. Hepatic steatosis in HCV core gene transgenic mice》
3.4. Hepatic steatosis in HCV core gene transgenic mice
Hepatic steatosis is more common in chronic hepatitis C than in chronic hepatitis B [40,48]. Indeed, steatosis was observed in 72% of chronic hepatitis C patients, versus only 19% of autoimmune chronic active hepatitis patients [49]. Hepatic steatosis is a reported risk factor for HCC in chronic hepatitis C [50]. Since the mechanism of hepatic steatosis is complicated due to the involvement of several metabolic pathways and interconnections with extrahepatic organs, the generation of transgenic mice carrying HCV proteins has been useful in assessing the direct contribution of HCV to the development of hepatosteatosis.
Among the transgenic mice that have been generated for HCV proteins, only core gene mice exhibited spontaneous hepatic steatosis. The LDs were initially small in young mice, with large droplets becoming predominant with age to resemble the livers of chronic hepatitis C patients. All male and approximately half of female transgenic mice exhibited hepatic steatosis by the age of 6 months [40]. Hepatic insulin resistance and steatosis are features shared by both HCV-infected patients and HCV core gene transgenic mice, while lymphocyte infiltration into hepatic parenchyma and bile duct injury are observed in HCV-infected patients alone [48].
HCV core protein-induced non-obese hepatosteatosis is associated with hypoadiponectinemia and can be improved by the administration of adiponectin [51]. PA28 is not only related to insulin resistance via TNF-α overproduction, but also involved in FA synthesis in HCV core gene transgenic mice, since the HCV core protein activates the sterol regulatory element-binding protein 1 promoter in a liver X receptor α/retinoid X receptor α (RXRα)- and PA28-dependent manner [52–55].
The influence of the HCV genotype 1b and 3a core proteins on the FA synthase (FAS) promoter and the molecular mechanism behind this process has been described. Microsomal triglyceride transfer protein (MTP) can promote the synthesis of very-lowdensity lipoprotein (VLDL). Since the HCV core protein lowers MTP activity [56], this function of the core protein on MTP may cause the accumulation of triglycerides [57]. It has also been demonstrated that the HCV core protein can directly bind to the RXRα–peroxisome proliferator-activated receptor α (PPARα) complex, a nuclear receptor governing FA β-oxidation, to activate signaling. Persistent PPARα–RXRα activation by the HCV core protein and excessive FAs in hepatocytes have been shown to enhance oxidative stress [58–60]. Moreover, a study has shown that HCV infection inhibits autophagy in hepatocytes [61]. PA28 activation leads to the degradation of microtubule-associated proteins 1A/1B light chain 3 (LC3)-I proteasomes, thereby preventing autophagy and enhancing lipid storage [62,63]. As HCV core gene transgenic mice have mitochondrial dysfunction, presumably due to the direct effect of the HCV core protein [64], this vicious cycle exacerbates mitochondrial damage and FA accumulation in the liver [57,60,65]. The possible mechanisms of steatogenesis by the HCV core protein are summarized in Fig. 3.
《3.5. HCC in HCV core gene transgenic mice》
3.5. HCC in HCV core gene transgenic mice
The HCV core protein also plays a prominent role in the development of HCC caused by chronic HCV infection. The incidence of HCC in HCV core protein transgenic mice (C21 and C49) was significantly higher than that in normal controls [16]. HCV core transgenic mice displayed clinicopathological characteristics similar to those of patients with chronic HCV infection, including a high frequency of accompanying steatosis [48], increased accumulation of carbon 18 monounsaturated FAs in the liver [66], mitochondrial dysfunction [38], increased insulin resistance and oxidative stress [67], and multicentric HCC occurrence [58,68,69]. The incidence of HCC was lower in female transgenic mice, which was consistent with epidemiological data showing that men are more likely to experience HCC among chronic hepatitis C patients [2,16,70]. Liver nodules with the characteristics of hepatocellular adenoma were found in 16-month-old HCV core transgenic mice. Fat droplets were abundant in the cytoplasm of hepatocellular adenoma cells, as in the cytoplasm of non-tumorous hepatocytes, with few droplets in HCC cells. This phenomenon in HCV core gene transgenic mice closely resembled that found in chronic hepatitis C patients, in which precancerous and well-differentiated HCC lesions occasionally showed marked fatty changes and reduced fat content during malignant transformation [16]. However, HCV core transgenic mice did not display liver inflammation or fibrosis during the hepatocarcinogenic process, in contrast to patients with chronic HCV infection [40,71].
The HCV core protein modulates the occurrence of HCC through multiple pathways. For example, long-term PPARα activation upregulated multiple oncogenic factors, including c-Fos, c-Myc, cyclin D1, cyclin-dependent kinase 4, proliferating cell nuclear antigen, and phosphorylated extracellular signal-regulated kinase (ERK) in rodents [58,72–74]. The HCV core protein can constitutively activate activator protein-1 (AP-1), which is related to the activation of c-Jun N-terminal kinase (JNK) and mitogenactivated protein kinase (MAPK) [53,75,76], while downregulating the activity of the tumor suppressor genes p53 and p21 [77].
Oxidative and ER stress are important factors in cell injury and drive malignant transformation. PPARα activation increased reactive oxygen species (ROS)-generating enzymes, such as acyl-CoA oxidase and cytochrome P450 4A1, which might cause nuclear DNA damage [58]. It has been reported that the HCV core protein triggered a calcium deficiency and stress in the ER, leading to the activation of caspase and BAX [78,79]. Persistent ER stress may also result in genomic instability and mutation and resistance to cell death [80]. These events may drive malignant transformation and aberrant cell fate, thus accelerating the progression of HCC [81] (Fig. 4).
《Fig. 4》

Fig. 4. Mechanism of HCC induced by the HCV core protein. The HCV core protein induces HCC in transgenic mice via multiple pathways. Oxidative and ER stress drive malignant transformation. The activation of PPARα–RXRα upregulates c-Myc, c-Fos, ERK, and cyclin D1 to induce aberrant cell proliferation. Furthermore, the activation of NF-κB, AP-1, JNK, STAT, and MAPK pathways, as well as hyperinsulinemia, affects cell proliferation. The combination of the above factors promotes hepatocarcinogenesis.
《4. Impact of lifestyle and dietary habits on the development of steatosis-derived HCC in HCV core gene transgenic mice》
4. Impact of lifestyle and dietary habits on the development of steatosis-derived HCC in HCV core gene transgenic mice
Several epidemiological studies have demonstrated that lifestyle and dietary factors can affect the clinical course of chronic hepatitis C patients [82–84]. In 9221 patients with chronic hepatitis C during 13.3 years of follow-up, participants who reported a diet high in protein had a significantly higher risk of hospitalization or death due to cirrhosis or liver cancer after adjusting for potential confounders [82]. Although total fat consumption was not remarkably associated with the risk of cirrhosis or liver cancer, a significant relationship was found for cholesterol consumption [82]. However, due to the complexity of lifestyle and dietary habits, it remains difficult to clarify which dietary features contribute to a diminished outcome and the precise mechanisms involved. To address these issues, comparisons between normal feeding and dietary intervention regimens in HCV core gene transgenic mice have been useful [8–11]. Meanwhile, non-alcoholic fatty liver disease (NAFLD)-derived HCC is increasing worldwide, with some HCC cases developing in steatotic livers without advanced fibrosis or cirrhosis [85,86]. Investigations of HCV core gene transgenic mice as a model of steatosis-derived HCC will provide clues for assessing how dietary interventions are harmful or useful for HCC associated with NAFLD. In this section, we summarize the impact of high-fat diets, a high-iron diet, ethanol, and dietary restriction on steatosis-derived hepatocarcinogenesis.
《4.1. Saturated fatty acid-rich diet》
4.1. Saturated fatty acid-rich diet
Saturated fatty acids (SFAs) are typical FAs that are abundantly found in animal products (e.g., red meat and dairy products) and plant products (e.g., palm oil and coconut oil) [87]. Excessive SFA intake is associated with obesity, insulin resistance, NAFLD, nonalcoholic steatohepatitis (NASH), and HCC [88–90]. When HCV core protein transgenic mice were treated with a control diet or an isocaloric SFA-rich diet for 15 months, macrovesicular LDs were more numerous in the SFA-treated group regardless of similar calorie intake [9]. Aggravation of hepatic steatosis by the SFA-rich diet was mainly due to the enhancement of FA synthesis in hepatocytes via the lipogenic enzymes acetyl-CoA carboxylase (ACC) α and β, FAS, and stearoyl-CoA desaturase 1 (SCD1). The prevalence of liver tumors increased significantly with the SFA-rich diet. Although there was no obvious hepatic fibrosis in the SFA-treated transgenic mice [9] (Table 1 [8–11,91,92]), the activation of NF-κB due to Tolllike receptor 4 (TLR4) and inflammasome signaling, JNK/AP-1 activation, induction of cyclin D1, and upregulation of the p62–nuclear factor erythroid 2-related factor 2 (NRF2) axis were presumably associated with the promotion of liver tumors [9].
《Table 1》
Table 1 Impact of lifestyle interventions on hepatic tumorigenesis in HCV core gene transgenic mice.
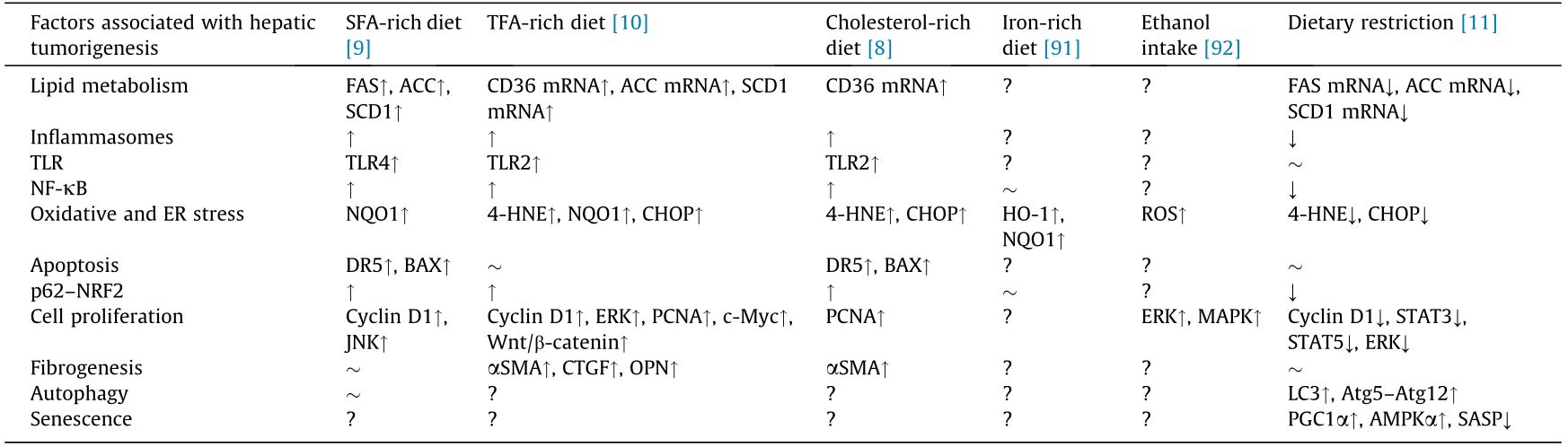
↑: Upregulated; ↓: downregulated;  : unchanged; ?: undetermined.
: unchanged; ?: undetermined.
HNE: hydroxynonenal; SMA: smooth muscle actin; AMPK: adenosine 5' -monophosphate-activated protein kinase; Atg: autophagy related; CD36: cluster of differentiation 36; CHOP: CCAAT/enhancer binding protein homologous protein; CTGF: connective tissue growth factor; DR: death receptor; HO: heme oxygenase; NQO: nicotinamide adenine dinucleotide/nicotinamide adenine dinucleotide phosphoric acid dehydrogenase; NRF: nuclear factor erythroid 2-related factor; OPN: osteopontin; PCNA: proliferating cell nuclear antigen; PGC: PPAR coactivator; SASP: senescence-associated secretory phenotype; TLR: Toll-like receptor.
《4.2. Trans fatty acid-rich diet》
4.2. Trans fatty acid-rich diet
Trans fatty acids (TFAs) are naturally present at low levels in dairy products and animal meat, while industrially generated TFAs are contained in hardened vegetable fats such as margarine and shortening, as well as in snack foods and fried foods. The WHO estimates that excessive TFA consumption leads to more than 500 000 deaths yearly from cardiovascular disease, and has called on governments to promote the REPLACE project in order to eliminate dietary TFAs [93]. Excessive TFA intake is associated with not only cardiovascular disease, but also shorter life expectancy, NAFLD, and cognitive disorders [94–96]. Earlier, we investigated the impact of a TFA-rich diet on hepatic tumorigenesis in HCV core gene transgenic mice [10]. Compared with the control diet group, TFA-rich diet-fed mice had significantly higher tumor prevalence. The TFA-rich diet significantly increased oxidative and ER stress, as evidenced by elevated levels of 4-hydroxynonenal (4-HNE), nicotinamide adenine dinucleotide/nicotinamide adenine dinucleotide phosphoric acid dehydrogenase 1 (NQO1), and CCAAT/enhancer binding protein homologous protein (CHOP). Increased oxidative and ER stress damage DNA and contribute to liver tumorigenesis. The TFA-rich diet also stimulated TLR2 and inflammasome signaling, activated NF-κB and p62–NRF2, and promoted fibrogenesis. Subsequently, the diet upregulated cyclin D1, ERK, c-Myc, Wnt/β-catenin signaling, and proliferating cell nuclear antigen (PCNA), driving the uncontrollable proliferation of transformed cells [10] (Table 1).
《4.3. Cholesterol-rich diet》
4.3. Cholesterol-rich diet
Animal-derived foods contain various amounts of cholesterol. The main dietary sources of cholesterol include red meat and egg yolks [97]. While cholesterol is essential for constructing cell membranes [98], its excessive intake is related to not only atherosclerosis and cerebrocardiovascular diseases, but also chronic hepatitis C progression [83,98–100]. Indeed, a cohort study using data from the hepatitis C antiviral long-term treatment against cirrhosis (HALT-C) project revealed elevated cholesterol intake to be associated with diminished clinical outcomes (death, variceal bleeding, hepatic decompensation, peritonitis, and HCC) [83]. When HCV core protein gene transgenic mice were fed a cholesterol-rich diet for 15 months, all of the mice experienced HCC [8]. The expression of cluster of differentiation 36 may have been related to steatosis caused by cholesterol in HCV core protein transgenic mice. Moreover, a cholesterol-rich diet induced NASH with pericellular fibrosis, promoted liver cell division, upregulated cellular stress, activated NF-κB and p62–NRF2, and ultimately accelerated liver tumorigenesis [8] (Table 1). These results helped in understanding the results of HALT-C: that higher cholesterol consumption was associated with disease progression in HCV-infected patients.
《4.4. Iron-rich diet》
4.4. Iron-rich diet
Iron is an essential auxiliary factor for life [101]. Since the element may generate free radicals through the Fenton reaction, it is reasonable to presume that excessive iron accumulation may increase cellular damage [91,102,103]. Indeed, hepatic iron accumulation was associated with the progression of chronic hepatitis C, and iron-depleting therapies such as phlebotomy attenuated disease-related hepatocyte damage [103,104]. When mice were fed a normal diet for 15 months, the hepatic iron content was significantly higher in HCV core gene transgenic mice as compared with normal mice [91]. After 3 months of an iron-rich diet, HCV core gene transgenic mice had significantly increased intrahepatic ROS levels in comparison with the control mice. The induction of anti-oxidant enzymes, such as heme oxygenase-1 (HO-1) and NQO1, was inhibited in transgenic mice, and diminishment of the iron-induced augmentation of HO-1 was confirmed in HepG2 cells expressing the core protein. This attenuation was not dependent on NRF2. Since enhanced oxidative stress may lead to nuclear DNA damage and promote the occurrence of HCC [91] (Table 1), it will be important to evaluate whether a long-term dietary iron overload promotes hepatic tumorigenesis in this transgenic mouse line in the future.
《4.5. Ethanol》
4.5. Ethanol
Ethanol is the main cause of pathogenesis in chronic liver disease, eventually leading to steatosis, steatohepatitis, fibrosis, and HCC [105]. The oxidative metabolism of ethanol disrupts signal transduction pathways and hampers the transcriptional control of some genes [106]. In HCV-infected patients, ethanol consumption was found to be significantly associated with the risk of HCC [107]. When a diet containing 5% ethanol was administered for 3 weeks, intrahepatic ROS levels increased significantly and ERK and p38 MAPK were activated in HCV core transgenic mice [92] (Table 1). Such findings strongly indicated that persistent ethanol intake promoted liver tumorigenesis in those mice. Further investigations are required to verify this hypothesis.
《4.6. Dietary restriction》
4.6. Dietary restriction
In recent years, calorie restriction has received considerable attention for health promotion and disease control. This method has been shown to have a preventive effect on obesity, metabolic syndrome, T2DM, and the progression of NAFLD/NASH, and is expected to be used to prevent and treat cancer in the future [108–111]. Earlier, we investigated whether dietary restriction could avert steatosis-associated liver tumorigenesis in HCV core protein transgenic mice [11]. Restricting the amount of food to 70% of a normal diet improved hepatic steatosis and significantly reduced the prevalence of liver tumors. Dietary restriction also markedly decreased hepatic oxidative and ER stress, significantly inhibited NF-κB activity and the expression of proinflammatory cytokines and senescence-associated secretory phenotypes, downregulated growth signaling via STAT3, STAT5, ERK, and insulin–Akt pathways, and activated autophagy [11] (Table 1). Indeed, dietary restriction may be a promising intervention to prevent NAFLDassociated hepatocarcinogenesis through multiple beneficial effects.
《5. Conclusions》
5. Conclusions
Techniques to generate transgenic mice expressing HCV proteins have opened new gateways in the fields of hepatology and virology by partially reproducing the phenotypes of HCV-infected patients and revealing the direct pathogenicity of HCV proteins in vivo. Only HCV core gene transgenic mice show hepatic insulin resistance, hepatic steatosis, and HCC and thus serve as a good model for determining the molecular events in steatosisassociated hepatocarcinogenesis. This mouse line illustrates the importance of a metabolic approach in the pathological analysis of liver disease. In addition, sequential studies using HCV core gene transgenic mice have shed light on the impact of dietary interventions on steatosis-derived HCC, which may be applicable to NAFLD-associated HCC in humans. HCV core gene transgenic mice will bolster the discovery of preventive agents against steatosisderived HCC. This valuable animal model straddles the intersection of virology, hepatology, metabolism, and nutrition, and highlights the importance of new engineering developments to bring about innovative discoveries in medical research.
《Acknowledgments》
Acknowledgments
The authors thank Mr. Trevor Ralph for his English editorial assistance.
This review has partially supported by JSPS Grants-in-Aid for Scientific Research (C) (KAKENHI; 16K08616, 16K08734, and 19K07383).
《Compliance with ethics guidelines》
Compliance with ethics guidelines
Pan Diao, Fangping Jia, Xiaojing Wang, Xiao Hu, Takefumi Kimura, and Takero Nakajima declare that they have no conflict of interest or financial conflicts to disclose.











 京公网安备 11010502051620号
京公网安备 11010502051620号




