
《1. Introduction》
1. Introduction
As the leading cause of death from a single infectious agent, Mycobacterium tuberculosis (M. tuberculosis) caused 10 million infections and 1.45 million deaths in 2018 worldwide [1]. The current recommended treatment against drug-susceptible tuberculosis (TB) requires a prolonged six-month combination therapy involving four first-line drugs. Furthermore, the rapid rise of multi-drug-resistant and totally drug-resistant TB renders this disease even more difficult to cure [2,3]. Thus, new therapeutic strategies for rapid and effective treatment of TB are highly desirable.
The development of new therapeutic methods against M. tuberculosis infections can be drastically accelerated by facile, precise, and markless genetic manipulation methods that allow for rapid identification and characterization of new drug targets in M. tuberculosis. Current methods for genetic manipulation in M. tuberculosis include allelic exchange using long linear DNA fragments [4], specialized transduction based on mycobacteriophages [5], recombineering mediated by a phage-encoded recombination system [6], oligonucleotide-mediated recombineering followed by Bxb1 integrase targeting [7], and non-homologous end joining (NHEJ) induced by highly specific double-strand breaks [8]. These methods either use multiple transformation steps and thus require months to years for editing, or can only generate non-precise mutations and thus are not amenable for gene function exploration at a single-base resolution.
Recently, clustered regularly interspaced short palindromic repeats (CRISPRs) and CRISPR-associated proteins (Cas) have been engineered for rapid and precise genetic manipulation in a variety of organisms, including many eukaryotic cells and diverse bacterial species [9–17]. Through base pairing with a genomic sequence adjacent to a protospacer adjacent motif (PAM), the Cas/guide RNA (gRNA) complexes can specifically bind to the target genomic site and generate a double-stranded DNA break (DSB). With the assistance of the cellular homology-directed repair (HDR) pathway or the NHEJ pathway, precise and non-precise genetic manipulations, respectively, can be readily achieved. Moreover, the CRISPR-Cas systems can be further engineered into transcription inhibition (CRISPRi) systems for direct gene knockdown without relying on cellular DNA repair mechanisms [18,19]. In M. tuberculosis, CRISPR-assisted NHEJ [8] and CRISPRi [20–23] strategies that allow for non-precise editing and partial gene knockdown, respectively, have been developed for genetic manipulation. However, although they are highly desirable, precise and complete gene inactivation methods are currently unavailable, likely due to the lack of a compatible HDR system.
More recently, the development of cytosine base editors (CBEs) and adenine base editors (ABEs) have provided new strategies for precise genetic manipulation [24–26]. Base editors composed of the fusion of a nucleotide deaminase and a dead Cas9 or a Cas9 nickase can directly achieve precise C-to-T or A-to-G conversions by means of a catalytic deamination reaction without requiring HDR; thus, they are widely applicable for precise genetic manipulation in a variety of microbes with different genetic backgrounds [27–35]. However, no such systems are currently available in M. tuberculosis.
In this study, we developed and characterized CTBE and CGBE in Mycobacterium smegmatis (M. smegmatis), and then applied CTBE for genome editing in M. tuberculosis. By screening distinct CRISPR base editors, we determined that only the unusual Streptococcus thermophilus Cas9 (St1Cas9) CBE—and not the widely used Streptococcus pyogenes Cas9 (SpCas9) or Lachnospiraceae bacterium Cpf1 (LbCpf1) CBEs—are active in M. smegmatis. Through systematic engineering of St1Cas9 CBE by means of uracil DNA glycosylase inhibitor (UGI) or uracil DNA glycosylase (UNG) fusion and the PAM expansion of St1Cas9, we created a C-to-T base editor and a C-to-G base editor with enhanced editing product purity, broadened targeting scope, and multiplexed editing capacity. Moreover, we evaluated the off-target effect of the C-to-T base editor by whole-genome sequencing and did not observe detectable offtarget editing events in M. smegmatis. Our approaches require only a single plasmid and one transformation step for efficient and scarless editing, significantly reducing the efforts and time required for precise genetic manipulation in mycobacterium.
《2. Materials and methods》
2. Materials and methods
《2.1. Bacterial strains and cultivation conditions》
2.1. Bacterial strains and cultivation conditions
The strains used in this study are listed in Table S1 in Appendix A, and the reagents used in this study are listed in Table S2 in Appendix A. The Escherichia coli (E. coli) TOP10 was used for plasmid construction and was cultured in Luria-Bertani (LB) broth at 37 °C. M. tuberculosis strain H37Rv, M. smegmatis strain mc2 155, and their derivative strains were used in this study. M. tuberculosis and M. smegmatis were grown at 37 °C in Middlebrook 7H9 broth or 7H10 plates supplemented with 0.2% glycerol, 0.05% Tween 80, 1× albumin dextrose catalase (ADC) (M. smegmatis), or oleic acid ADC (OADC) (M. tuberculosis) and the appropriate antibiotics. When noted, antibiotics or chemicals were used at the following concentrations: kanamycin, 20 μg∙mL–1 for M. smegmatis and M. tuberculosis, and 50 μg∙mL–1 for E. coli TOP10; leucine (50 μg∙mL–1 ); isoniazid (INH, 16 μg∙mL–1 ); and anhydrotetracycline (ATc, 20 ng∙mL–1 ).
《2.2. Plasmid construction》
2.2. Plasmid construction
The primers used for plasmid construction are listed in Table S3 in Appendix A, and the plasmids used in this study are listed in Table S4 in Appendix A.
2.2.1. Constructions of CBE_dSt1Cas9, CBE_dSpCas9, and CBE_dLbCpf1 plasmids
The J23119-driven single guide RNA (sgRNA)-expression cassette was synthesized by GENEWIZ (China), and was assembled into the backbone of the pLJR962 [22] plasmid (linearized by Esp3I and SapI) using T4 DNA ligase. The APOBEC1 gene was polymerase chain reaction (PCR)-amplified from pBECKP [33] and cloned into the backbone of the pLJR962 plasmid via Gibson assembly [36], resulting in the CBE_dSt1Cas9 plasmid. The APOBEC1 and dSt1Cas9 genes were linked by a 32 AA linker, and the expression of the APOBEC1–dSt1Cas9 fusion protein was under the control of the ATc-inducible promoter Ptet. The CBE_dSpCas9 and CBE_dLbCpf1 plasmids were constructed using a similar strategy to that used in the construction of the CBE_dSt1Cas9 plasmid.
2.2.2. Construction of the pMF1_CBE_dSt1Cas9, pJAZ38_CBE_dSt1Cas9, and pAL5000_CBE_dSt1Cas9 plasmids
The pMF1 replicon and Tet Repressor protein (TetR) gene were amplified from pYC1640 and assembled into the backbone of the pLJR962_CBE_dSt1Cas9 plasmid via Gibson assembly, resulting in the pMF1_CBE_dSt1Cas9 plasmid. Similar strategies were used to construct the pJAZ38_CBE_dSt1Cas9 and pAL5000_CBE_dSt1Cas9 plasmids.
2.2.3. Construction of the pMF1_CTBE_cons, pMF1_CGBE_cons, pMF1_CTBEengineer_cons, and pMF1_CGBEengineer_cons plasmids
UGI was synthesized by Sangon (China) and was assembled into the backbone of the pJLR962_CBE_dSt1Cas9 plasmid via Gibson assembly. The pMF1 replicon and TetR were amplified from pYC1640 and assembled into the aforementioned plasmid, resulting in the pMF1_CTBE_cons plasmid. E. coli UNG (eUNG) was amplified from the genome of E. coli MG1655 and assembled into the backbone of the pMF1_CBE_dSt1Cas9 plasmid via Gibson assembly, resulting in the pMF1_CGBE_cons plasmid. D939K/E1057Q/N1081K/K1086L mutations were introduced into the pMF1_CTBE_cons plasmid via Gibson assembly, resulting in the pMF1_CTBEengineer_cons plasmid. APOBEC1–dSt1Cas9 (D939K/E1057Q/N1081K/K1086L) was amplified from pMF1_CTBEengineer_cons and assembled into the backbone of the pMF1_CGBE_cons plasmid, resulting in the pMF1_CGBEengineer_cons plasmid. These plasmids were used for base editing in M. smegmatis.
2.2.4. Construction of the pMF1_CTBEengineer plasmid
The Tet-driven sgRNA-expression cassette was introduced into pMF1_CTBEengineer_cons via Gibson assembly, resulting in pMF1_CTBEengineer. In this plasmid, both APOBEC1–dSt1Cas9–UGI and sgRNA were under the control of the ATc-inducible promoter Ptet. This plasmid was used for base editing in M. tuberculosis.
《2.3. Competent cell preparation and electroporation》
2.3. Competent cell preparation and electroporation
M. tuberculosis strain H37Rv (ATCC27294) was inoculated in Lowenstein-Jensen slant from frozen stock and incubated at 37 °C for 2 weeks; it was then transferred and grown in 100 mL of Middlebrook 7H9 broth supplemented with 0.05% Tween 80, 0.2% glycerol, and OADC for another 2 weeks. The culture was cooled on ice for 5 min and collected using centrifugation. The pellets were washed twice with 30 mL of 10% precooled glycerol and resuspended in 5 mL of 10% cold glycerol. For electroporation, 1 μg of recombinant plasmid was mixed with 100 μL of competent cells in a 0.2 cm cuvette; the transformation was conducted with the Gene Pulser Xcell Electroporation System (Bio-Rad, USA) under the following conditions: 2.5 kV, 1000 Ω, and 25 μF. After the shock, 1 mL of 7H9 broth supplemented with OADC was immediately added into the cuvette. The culture was incubated for 2 d at 37 °C and plated on Middlebrook 7H10 agar supplemented with OADC, 20 μg∙mL–1 kanamycin, and 20 ng∙mL–1 anhydrous tetracycline. The plates were sealed by parafilm and incubated for 20– 30 d at 37 °C.
M. smegmatis strain mc2 155 (ATCC700084) was inoculated in 7H10 with 10% ADC enrichment from frozen stock and incubated at 37 °C for 4 d. A single colony of a M. smegmatis strain was inoculated into 2 mL of Middlebrook 7H9 broth supplemented with 0.05% Tween 80, 0.2% glycerol, and ADC at 37 °C for 24 h. The cells were diluted 1:100 into 100 mL of Middlebrook 7H9 broth supplemented with 0.05% Tween 80, 0.2% glycerol, and ADC for another 12–15 h. When the optical density at 600 nm of the culture reached 0.8 to 1, the culture was cooled on ice for 20 min and collected using centrifugation in 50 mL conical tubes at 4000 r∙min–1 for 10 min. The pellets were washed twice with 30 mL of 10% precooled glycerol and resuspended in 10 mL of 10% cold glycerol. For electroporation, 100 ng of plasmid was mixed with 100 μL of competent cells in a 0.2 cm cuvette; the transformation was conducted with a Gene Pulser Xcell Electroporation System under the following conditions: 2.5 kV, 1000 Ω, and 25 μF. After the shock, 1 mL of 7H9 broth supplemented with ADC was immediately added into the cuvette. The culture was incubated for 3 h at 37 °C, and 10% portions of the culture were plated on Middlebrook 7H10 agar supplemented with ADC, 20 μg∙mL–1 kanamycin, and 20 ng∙mL–1 anhydrous tetracycline. The plates were sealed with parafilm and incubated for 7–10 d at 37 °C.
《2.4. Editing efficiency evaluation》
2.4. Editing efficiency evaluation
The sgRNA target sequences used in this study are listed in Table S5 in Appendix A. After transformation, the plates were sealed with parafilm and incubated at 37 °C. All colonies of M. smegmatis or M. tuberculosis were collected from the plates, and the genomic DNAs were extracted using a Rapid Bacterial Genomic DNA Isolation Kit (Sangon Biotech). The target region was amplified with Easy Taq DNA Polymerase (TransGen, China), using specific primers for the target region. The PCR products were sent out for Sanger sequencing, and the editing efficiency was calculated by using EditR 1.0.10 [37].
《2.5. leuB or leuC knockout using CTBEengineer》
2.5. leuB or leuC knockout using CTBEengineer
Spacers were designed and inserted into the CTBEengineer plasmid. The successfully constructed plasmids were then electroporated into M. smegmatis mc2155 competent cells. After the shock, 1 mL of Middlebrook 7H9 broth with ADC and leucine was immediately added into the cuvette. The culture was incubated for 3 h at 37 °C, and 10% portions of the culture were plated onto a Middlebrook 7H10 agar plate supplemented with ADC, 20 μg∙mL–1 kanamycin, 20 ng∙mL–1 anhydrous tetracycline, and 50 μg∙mL–1 leucine. The plates were sealed with parafilm and incubated at 37 °C. Seven days after electroporation, the overall editing efficiencies were evaluated using the method described in Section 2.4. Single colonies were separately cultured in 5 mL of 7H9 broth supplemented with ADC and leucine at 37 °C for 2 d in the absence of kanamycin. The cells were then plated onto Middlebrook 7H10 agar plates supplemented with ADC and leucine. The leuB or leuC mutant strains were isolated and confirmed by sequencing.
《2.6. Plasmid curing》
2.6. Plasmid curing
To cure the editing plasmid in M. smegmatis after base editing, one colony was cultured in Middlebrook 7H9 broth with ADC in the absence of kanamycin. After growing to the stationary phase (4 d), the cells were plated onto a Middlebrook 7H10 agar plate supplemented with ADC. A single colony was picked and diluted into 5 mL of Middlebrook 7H9. A fraction of the diluted cells were plated onto a Middlebrook 7H10 agar plate supplemented with ADC without kanamycin, and another fraction was plated onto a Middlebrook 7H10 agar plate supplemented with ADC containing kanamycin. The cells whose plasmid was successfully cured could only grow on the plate without kanamycin.
《2.7. Leucine auxotrophy assay》
2.7. Leucine auxotrophy assay
The leuB or leuC gene was knocked out by introducing a premature stop codon using the CTBEengineer plasmid. The strains were grown in Middlebrook 7H9 broth supplemented with ADC and leucine to the stationary phase. A fraction of the cells were plated onto a Middlebrook 7H10 agar plate supplemented with ADC without leucine, and another fraction was plated onto a Middlebrook 7H10 agar plate supplemented with ADC and leucine. The plates were sealed with parafilm and incubated for 4 d at 37 °C.
《2.8. Isoniazid resistance assay》
2.8. Isoniazid resistance assay
The katG gene was knocked out by introducing a premature stop codon at Gln3 (CAA to TAA) using the CTBEengineer plasmid. The strains were grown in Middlebrook 7H9 broth with OADC to the stationary phase. A fraction of the cells were plated onto a Middlebrook 7H10 agar plate supplemented with OADC in the absence of INH, and another fraction was plated onto a Middlebrook 7H10 agar plate supplemented with OADC and INH (16 μg∙mL–1 ). The plates were sealed with parafilm and incubated for 20 d at 37 °C.
《2.9. Aggregation assay》
2.9. Aggregation assay
The ctpE gene was knocked out by introducing a premature stop codon at Gln16 (CAG to TAG) using the CTBEengineer plasmid. The strains were grown in Middlebrook 7H9 broth to the mid-log phase. The cells were diluted 1:100 and cultured in Middlebrook 7H9 broth containing 1.0 mmol∙L–1 ethylenebis (oxyethylenenitrilo) tetraacetic or 0 mmol∙L–1 EGTA. The cells were grown at 37 °C for 48 h with shaking at 200 r∙min–1 ; then, the cells were left undisturbed for 1.0 h at room temperature for cell aggregation.
《2.10. Whole-genome sequencing》
2.10. Whole-genome sequencing
Genomes of the wild-type (WT) and two edited strains were sent out for whole-genome sequencing by the Illumina HiSeq/Nova 2× 150 bp platform at GENEWIZ. The paired-end fragment libraries were sequenced according to the Illumina HiSeq/Nova 2× 150 bp platform’s protocol. The pass filter data was processed using cutadapt (v1.9.1) to obtain clean data, which was aligned with the reference genome (Accession number: NC_008596) using BWA (version 0.7.17). The output file was then processed using Picard and The Genome Analysis Toolkit (GATK) for duplicate removal, local realignment, and base quality recalibration. Singlenucleotide variants (SNVs) were detected using the Haplotype Caller module provided by GATK and rearranged using Excel (Microsoft, USA). The output SNVs were aligned with the potential off-target sites containing the identical sequences to the target sites at 1–8 nucleotide (nt) proximal to the PAM in order to assess the genome-wide off-target effect.
《2.11. Preparation of sgRNA》
2.11. Preparation of sgRNA
The transcription template (double-stranded DNA) of sgRNA was chemically synthesized by GENEWIZ, and the sgRNA template was amplified by PCR. sgRNA was transcribed in vitro using the HiScribe T7 High Yield RNA Synthesis Kit (NEB, USA) following the manufacturer’s instructions. After the amplified and transcribed template was being incubated at 37 °C overnight, deoxyribonuclease I (DNase I) was added to eliminate DNA templates. The products were further purified using phenol/chloroform extraction followed by ethanol precipitation. After purification, the sgRNA was stored at –80 °C.
《2.12. In vitro DNA cleavage assay for St1Cas9 proteins》
2.12. In vitro DNA cleavage assay for St1Cas9 proteins
The preparation of the proteins and the cleavage assay were performed by following a previous study [43]. In brief, the sequence containing the spacer and an AAAGAA PAM was first cloned into a pUC19-based vector (referred to as pUC19-AGAA). Plasmids with distinct PAM sites were constructed via sitespecific mutagenesis (Table S6 in Appendix A). The plasmids were linearized by means of KpnI digestion overnight, and the products were purified using the TIANquick Midi Purification Kit (TIANGEN, China) as the cleavage substrates. For the in vitro cleavage assay, 250 nmol∙L–1 of purified St1Cas9 or its variants was mixed with 500 nmol∙L–1 of sgRNA in the reaction buffer (10 mmol∙L–1 Tris-HCl (pH 7.5), 500 mmol∙L–1 NaCl, 1.5 mmol∙L–1 MgCl2, and 1 mmol∙L–1 dithiothreitol (DTT)). Next, linearized plasmids were added to the reaction buffer (for a final concentration of 5 nmol∙L–1 ). The reactions were incubated at 37 °C for 40 min and then transferred to liquid nitrogen immediately. Then, 25 mmol∙L–1 EDTA and 10 μg Proteinase K were added to the reaction tube to terminate the reaction. After incubation at 58 C for 10 min, the reaction products were analyzed using a 1% agarose gel. The products were stained with 4S Red Plus (Sangon Biotech) and visualized by the ChemiDoc MP System (Bio-Rad).
《3. Results and discussion》
3. Results and discussion
《3.1. Identification of active cytosine base editors in M. smegmatis》
3.1. Identification of active cytosine base editors in M. smegmatis
SpCas9 has been widely adapted for genetic manipulation in numerous microbes [16,29,38–41]. However, it is restricted for application in mycobacteria because of its notable cellular toxicity and low DNA-targeting efficiency [8,22]. To develop an active CBE in mycobacteria, we screened various CBEs composed of a fusion of rat APOBEC1 cytosine deaminase and different Cas nucleases, including dSpCas9 (catalytically inactive SpCas9), dLbCpf1 (catalytically inactive LbCpf1), and dSt1Cas9 (catalytically inactive St1Cas9). The expression of the Cas nucleases was under the control of an ATc-inducible promoter, while the expression of the corresponding gRNAs was under the control of a synthetic constitutive promoter J23119.
To quantitatively assess the efficiency of targeted base editing, the same amount of each of the different CBE plasmids was electroporated into M. smegmatis. CBEdSt1Cas9 (CBE_dSt1Cas9) induced notable C-to-T conversions (from 4% to 15%) with undesired Cto-G conversions (from 18% to 70%) as the major editing products at all the test sites, whereas the editing efficiency of CBEdSpCas9 (dSpCpf1 CBE) and CBEdLbCpf1 (dLbCpf1 CBE) was lower than 10% at all the target sites (Fig. 1(a)). Moreover, in line with previous studies [42,43], no notable cellular toxicity was observed for CBEdSt1Cas9 or CBEdLbCpf1, whereas high cellular toxicity was observed for CBEdSpCas9 (Fig. 1(b)). Therefore, we chose CBEdSt1Cas9 as the starting system for further engineering. To cure the plasmid after base editing, we replaced the non-replicating L5 integrating backbone used in the CBEdSt1Cas9 system with the replicable pMF1 backbone. We found that the resulting pMF1 backbone based CBEdSt1Cas9 plasmid did not show notable cellular toxicity and could be easily cured by culturing the bacterium in the absence of antibiotic (Figs. S1 and S2 in Appendix A).
《3.2. Development of CTBE and CGBE in M. smegmatis》
3.2. Development of CTBE and CGBE in M. smegmatis
The G:U mismatch pair in cells is generally repaired by the UNG-mediated base excision repair (BER) process. Inhibition of UNG would protect the edited G:U intermediate from cleavage and thus improve C-to-T conversion efficiency and editing product purity [24,44] (Fig. 1(c)). We fused two UGIs to the C terminus of dSt1Cas9 to create the C-to-T base editor (CTBEWT). To examine the C-to-T editing efficiency of CTBEWT, five CTBEWT plasmids targeting five different loci were separately electroporated into M. smegmatis. CTBEWT achieved high levels of C-to-T base editing frequency ranging from 69% to 86%, with significantly reduced formation of undesired byproducts (Fig. 1(d)).
Recent studies have revealed that promoting the BER pathway by means of UNG or other DNA repair proteins fusion to SpCas9 CBEs can convert a target C:G base pair into a G:C or A:T base pair, rather than the expected T:A product [45–48]. Given that a high proportion of unexpected C-to-G byproduct existed with CBEdSt1Cas9, we anticipated that fusion of UNG to CBEdSt1Cas9 would generate new base editors. We thus fused eUNG to the C terminus of dSt1Cas9 in CBEdSt1Cas9 to create a new base editor, CGBEWT, and compared the base editing efficiencies of CGBEWT and CBEdSt1Cas9. As shown in Fig. 1(e), CGBEWT can efficiently achieve C-to-G conversions with enhanced editing product purity at all five tested sites.
《Fig. 1》
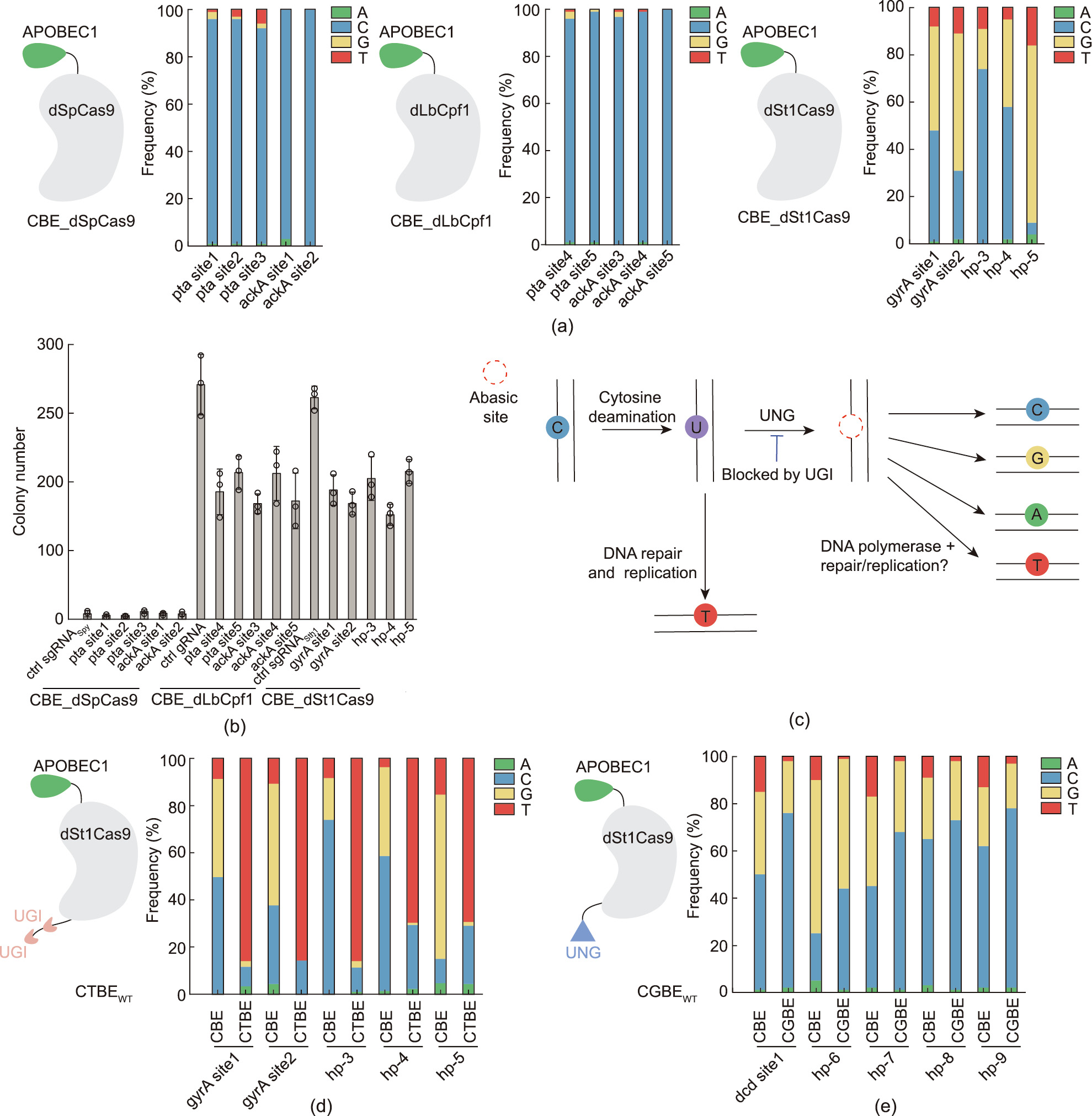
Fig. 1. St1Cas9-mediated base editing in M. smegmatis. (a) Identification of active CRISPR base editors in M. smegmatis. Three distinct base editors, S. thermophilus Cas9 CBE (CBE_dSt1Cas9), S. pyogenes Cas9 CBE (CBE_dSpCas9), and L. bacterium Cpf1 CBE (CBE_dLbCpf1), were screened. APOBEC1 cytosine deaminase was fused to the N terminus of the Cas nucleases via a 32 AA linker. (b) Transformation efficiencies of the distinct base editors in M. smegmatis; 100 ng of each plasmid was used for electroporation. (c) Possible cellular DNA repair mechanisms of cytosine deamination. The initial editing product, the U:G mismatch pair, can be directly converted into the T:A pair by means of DNA repair and replication or it can be excised by endogenous UNG, leading to the formation of diverse editing products. UGI can block endogenous UNG activity. (d, e) St1Cas9 CBE was engineered with UGI or UNG fusion, yielding two new base editors, (d) CTBE and (e) CGBE, capable of C-to-T or C-to-G conversions in M. smegmatis with drastically enhanced editing purity.
《3.3. Engineering of a PAM-expanded St1Cas9 variant for the base editors》
3.3. Engineering of a PAM-expanded St1Cas9 variant for the base editors
St1Cas9 requires a relatively strict PAM sequence (5' -NNRGAA3' , where R is A or G) for DNA targeting [43], significantly restricting the editing scope of CTBEWT and CGBEWT. We previously engineered a St1Cas9 variant, KLKL, by introducing D939K/E1057L/N1081K/K1086L mutations to relieve the PAM specificity [43]. In wild-type St1Cas9, Q1084 and K1086 from bidentate hydrogen bond with the third A and fourth G when 5' -NNAGGA-3' PAM is used, and Q1084 is further stabilized by E1057 via a hydrogen bond (Fig. 2(a)). Mutations of E1057L and K1086L would disrupt base-specific interactions, while mutations of D939K and N1081K would introduce non-base-specific interactions [43]. We recently found that mutation of L1057Q in the KLKL variant would increase the in vitro DNA cleavage activity toward 5' -NNTTAA-3' PAM- and 5' -NNCTAA-3' PAM-containing DNAs (Fig. 2(b)). We therefore engineered a St1Cas9 KQKL variant containing D939K/E1057Q/N1081K/K1086L mutations to further relieve the PAM specificity. A comprehensive in vitro DNA cleavage assay revealed that mutation of D939K/E1057Q/N1081K/K1086L substantially expanded the PAM-recognition scope of the KQKL variant with the 5' - NNNNAA-3' PAM specificity, compared with the 5' -NNRGAA-3' specificity of the WT St1Cas9 (Fig. 2(c)).
《Fig. 2》
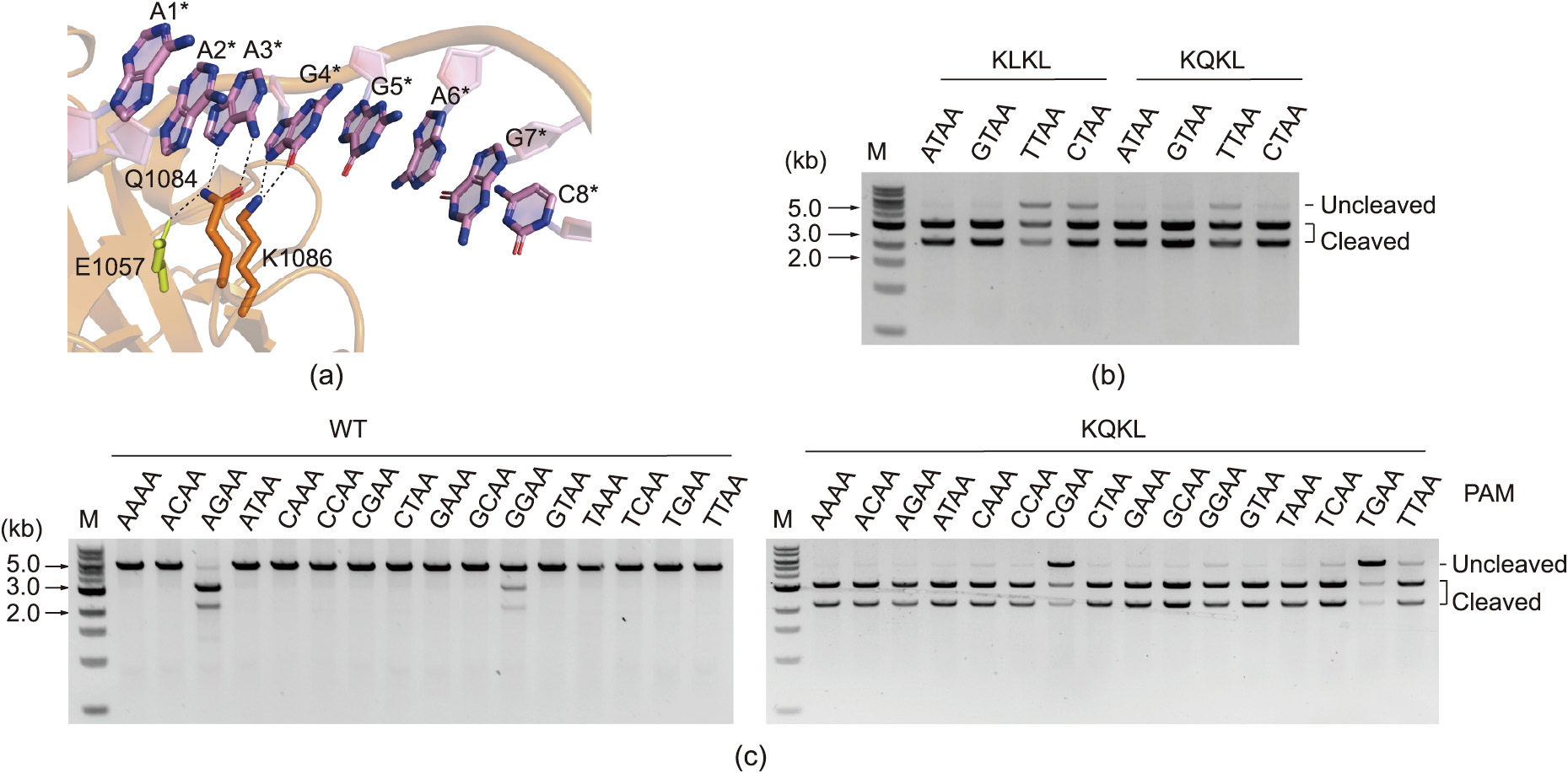
Fig. 2. PAM expansion of St1Cas9 via structure-guided engineering. (a) PAM-recognition mechanism of WT St1Cas9 (PDB: 6M0X). (b) In vitro DNA cleavage assay of two engineered mutants of St1Cas9. (c) In vitro DNA cleavage assay of WT St1Cas9 and the KQKL variant. M: marker.
Next, we replaced the WT St1Cas9 of CTBEWT with the KQKL variant to expand the editing scope of the C-to-T base editor (Fig. 3(a)). The resulting CTBEengineer system was first subjected to ATc concentration optimization, because ATc is the inducer for the expression of the engineered St1Cas9 variant. We screened six different concentrations of ATc, ranging from 5 to 100 ng∙mL–11 , but observed no significant differences in the editing efficiencies (Fig. S3 in Appendix A). We selected 20 ng∙mL–1 as the inducer concentration for base editing in the subsequent experiments.
To systemically characterize the PAM preference of CTBEengineer in vivo, we assembled 48 spacers targeting 48 different endogenous genomic sites, with three spacers for each 5' -NNNNAA-3' PAM. We collected all the colonies after each transformation and subjected the PCR products of the target sites for sequencing to evaluate the editing efficiencies. Consistent with the in vitro DNA cleavage assay, CTBEengineer had an expanded PAM preference with the 5' -NNNNAA-3' PAM specificity (Fig. 3(b)). We also noticed that, when targeting the sites with less-active PAMs, such as 5' -NNTAAA-3' , 5' -NNTCAA-3' , 5' -NNTGAA-3' , and 5' -NNTTAA-3' , the spacer sequence content could significantly affect the editing efficiency of CTBEengineer (Fig. 3(b)). Moreover, we analyzed the editing window of CTBEengineer, revealing that CTBEengineer preferred to edit Cs within the window from positions 4 to 12 (Fig. 3(c)). When the Cs were located outside the window from positions 4 to 12, but inside the window from 2 to 15, the Cs could sometimes still be edited with high efficiency (Fig. 3(c)).
《Fig. 3》
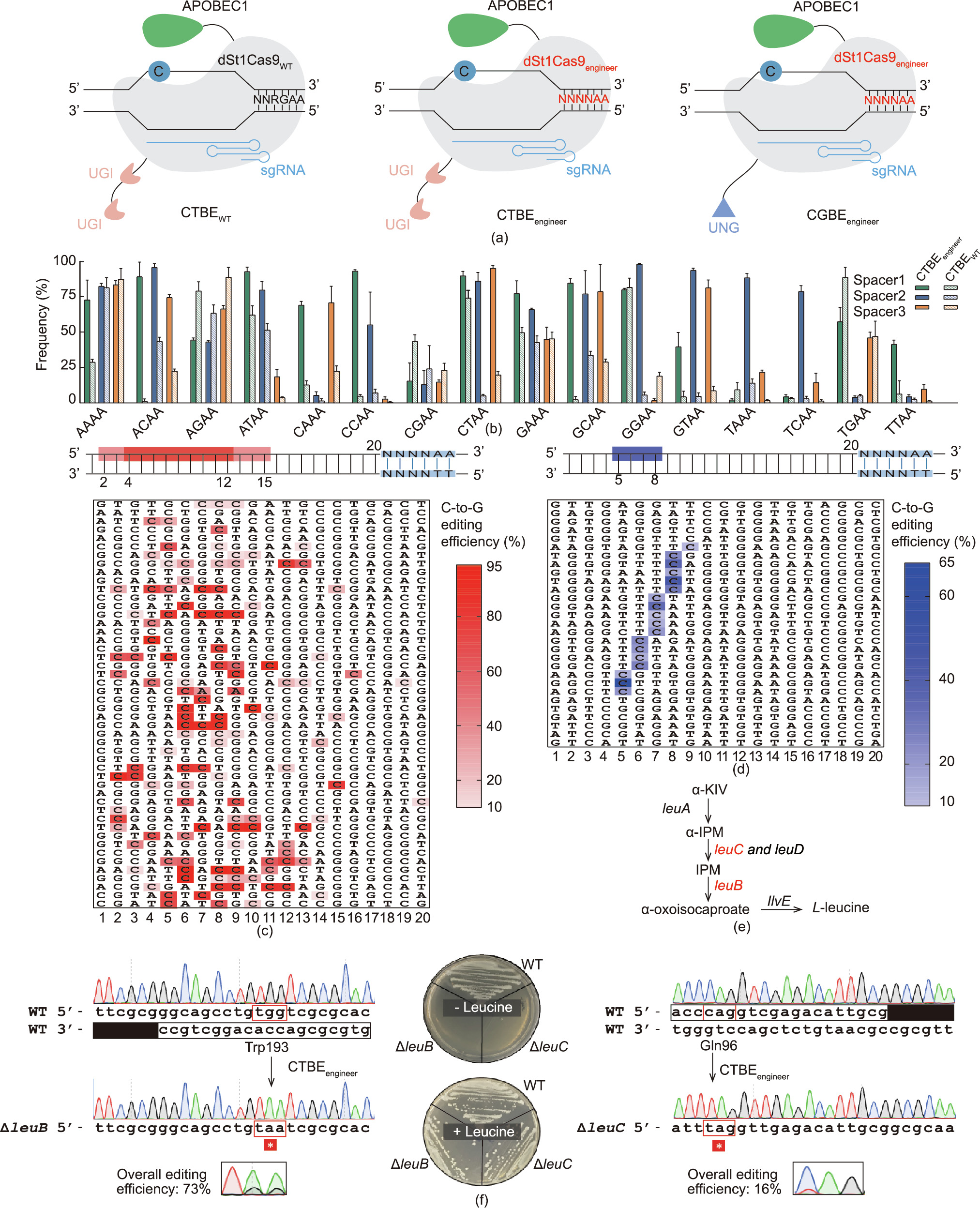
Fig. 3. Comprehensive characterizations of CTBE and CGBE in M. smegmatis. (a) Compositions of different St1Cas9 CBEs. (b) Editing activity comparison of CTBEWT and CTBEengineer in M. smegmatis (bar plots reflect the maximum editing frequency within the editing window). (c, d) Editing windows of (c) CTBEengineer and (d) CGBEengineer. (e) The L-leucine biosynthesis pathway in M. smegmatis. The targeted genes (leuC and leuB) are highlighted in red (α-KIV: α-ketoisovalerate; α-IPM: α-isopropylmalate; IPM: 3-isopropylmalate). (f) Inactivation of leuB and leuC via CTBEengineer by generating premature stop codons. Inactivation of leuB or leuC causes auxotrophy in the absence of L-leucine. A box and an inverted box indicate the target sequence and PAM, respectively.
Similarly, we engineered CGBEWT with St1Cas9engineer to construct the CGBEengineer system (Fig. 3(a)). We comprehensively characterized CGBEengineer by testing the editing efficiencies of 29 endogenous genomic sites. Interestingly, only the Cs within the window from positions 5 to 8 could be edited with the CGBEengineer system (Fig. 3(d)), although the same Cas9 protein and deaminase were used for CGBEengineer and CTBEengineer. Moreover, only Cs with the TC motif could be edited, even if they were located within the editing window from positions 5 to 8 (Fig. 3(d)).
In addition, we replaced the eUNG in CGBEengineer with an orthologous UNG from M. smegmatis (mUNG) to facilitate C-to-G conversions in M. smegmatis (Fig. S4 in Appendix A). Four versions of C-to-G base editors with the fusion of eUNG or mUNG to the C or N terminus of the St1Cas9 protein were constructed for base editing in M. smegmatis. We found that fusion of mUNG or eUNG to the C terminus of St1Cas9 gave similar editing efficiencies at all the tested sites, whereas fusion of mUNG or eUNG at the N terminus did not yield efficient C-to-G editing (Fig. S4). Given the similar editing efficiency using mUNG or eUNG, we kept eUNG in CGBEengineer for further characterization.
《3.4. Gene inactivation by CTBEengineer in M. smegmatis》
3.4. Gene inactivation by CTBEengineer in M. smegmatis
CRISPR C-to-T base editors can convert CAA, CGA, CAG, and TGG codons to premature stop codons; thus, they are promising tools for gene inactivation [49,50]. We examined the gene inactivation capacity of CTBEengineer by designing three different spacers targeting the essential L-leucine biosynthesis genes leuB and leuC. To evaluate the overall editing efficiency, the genomic DNA of all colonies on the plate were extracted, and the target regions were amplified and sequenced. Efficient editing was observed for all the designed spacers (Fig. 3(f) and Fig. S5 in Appendix A), and the isolated pure mutants were subjected to a phenotypical assay. Generating a premature stop codon in leuB or leuC rendered the bacterium incapable of growth in the absence of L-leucine, confirming the inactivation of L-leucine biosynthesis (Fig. 3(f)). Moreover, a similar strategy was successfully applied to inactivate ctpE by generating a premature stop codon with CTBEengineer. In line with previous studies [51], inactivation of ctpE (calcium-transporting adenosine 5' -triPhosphatase) by introducing the premature stop codon increased bacterium aggregation in the presence of EGTA (Fig. S6 in Appendix A). Together, these results confirmed that CTBEengineer is a powerful and reliable tool for gene inactivation in M. smegmatis.
《3.5. Multiplexed editing in M. smegmatis》
3.5. Multiplexed editing in M. smegmatis
Serial genome editing for multiple genes in slow-growing pathogens is extremely time consuming, and multiplexed genome editing can drastically expedite the genome-editing progress. We assembled two sgRNA-expression cassettes into a single CTBEengineer plasmid to test the multiplexed editing capacity of CTBEengineer in the M. smegmatis mc2155 strain (Fig. S7(a) in Appendix A). As shown in Fig. S7(b) in Appendix A, two genes (sigF and Ms6753) were simultaneously mutated in six out of eight randomly picked colonies. Moreover, three different genomic sites (cysS, sigF, and Ms6753) were targeted simultaneously by assembling three sgRNAs into the CTBEengineer plasmid, and all three targeted sites were successfully mutated in seven out of eight randomly picked colonies (Figs. 4(a, b)). Similarly, we assembled two or three sgRNA-expression cassettes into a single CGBEengineer plasmid and tested the multiplexed editing capacity of C-to-G conversion in the M. smegmatis mc2155 strain (Fig. S8(a) in Appendix A and Fig. 4(c)). As shown in Fig. S8(b) in Appendix A, CGBEengineer succeeded in double mutagenesis in six out of the eight analyzed clones. For the triple mutagenesis assay, all three targeted sites were successfully mutated in six out of eight analyzed colonies (Fig. 4 (d)). Together, these results demonstrate that both CTBEengineer and CGBEengineer are amenable for multiplexed editing in M. smegmatis.
《Fig. 4》
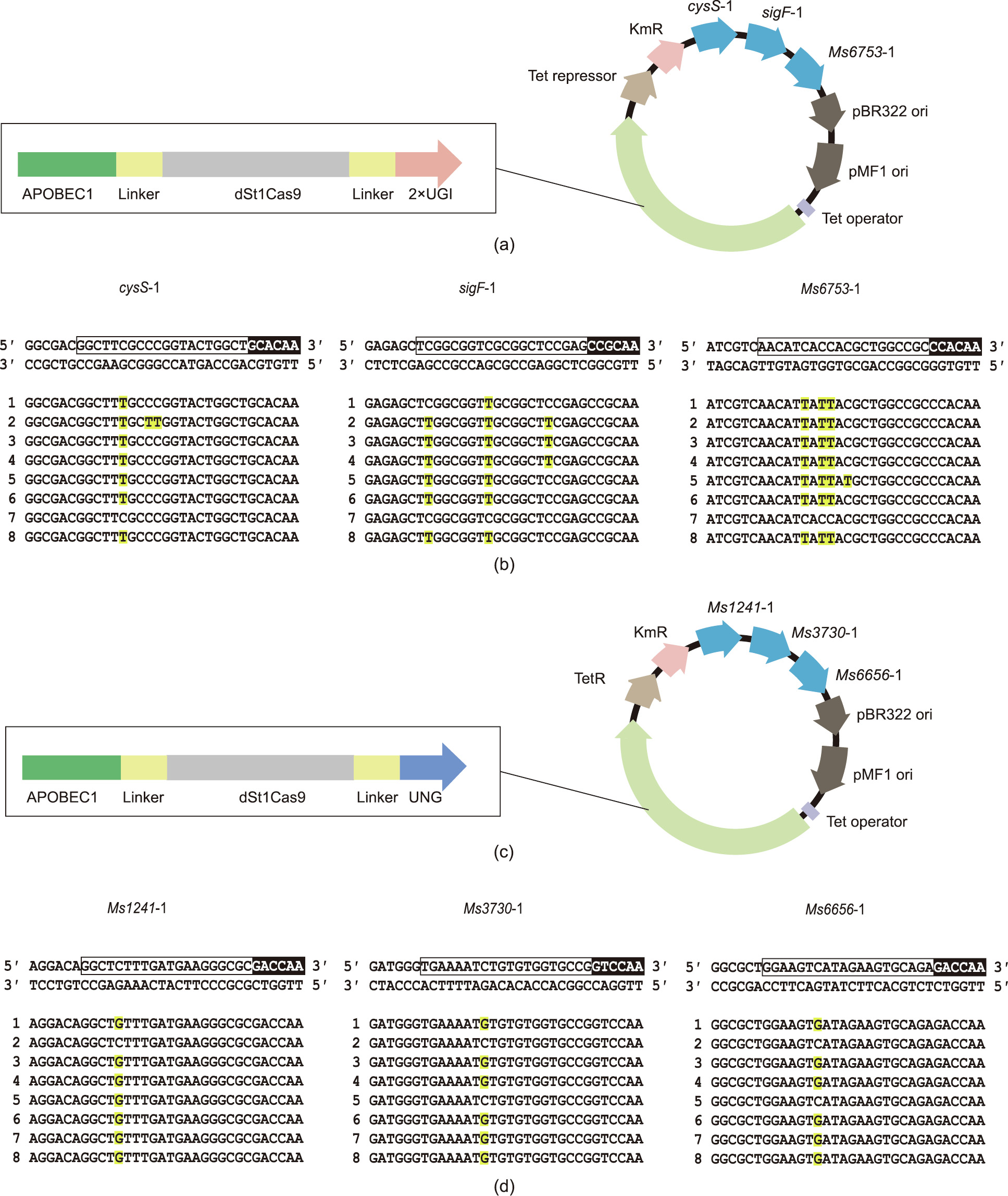
Fig. 4. Multiplexed editing in M. smegmatis. (a) Map of the single-plasmid system for CTBEengineer-mediated multiplexed mutagenesis. (b) Editing results of the CTBEengineer-mediated multiplexed editing assay. A box and an inverted box indicate the target sequence and PAM, respectively. The edited bases are shown in red and highlighted in yellow. (c) Map of the single-plasmid system for CGBEengineer-mediated multiplexed mutagenesis. (d) Editing results of the CGBE-mediated multiplexed editing assay. A box and an inverted box indicate the target sequence and PAM, respectively. The edited bases are shown in red and highlighted in yellow.
《3.6. Genome-wide off-target evaluation for CTBEengineer》
3.6. Genome-wide off-target evaluation for CTBEengineer
To evaluate the genome-wide off-target editing of the CTBEengineer system, two edited M. smegmatis colonies were randomly selected and subjected to whole-genome sequencing. No potential off-target editing sites containing sequences identical to the PAM proximal 1–8 nt of the protospacer were detected (Table S7 in Appendix A). These results demonstrate the high editing fidelity of the CTBEengineer system and are consistent with previous discoveries that St1Cas9 is a high-fidelity enzyme and its editing is highly sensitive to mutations in the spacer sequences [43].
《3.7. Genome editing in M: tuberculosis》
3.7. Genome editing in M: tuberculosis
Given the success of base editing with CTBEengineer in M. smegmatis, we sought to examine the base editing capacity of this system in M. tuberculosis. Four different spacers targeting four different endogenous sites were separately cloned into the CTBEengineer system, and the resulting editing plasmids were separately electroporated into the M. tuberculosis H37Rv competent cells. C-to-T conversion efficiencies were measured by collecting all the transformants and sequencing the target sites. Notable C-to-T conversions were observed for all four tested sites, with the editing frequencies ranging from 12% to 95% (Fig. 5(a)). Moreover, we applied this system for gene inactivation by introducing a premature stop codon into katG (Fig. 5(b)). Inactivation of katG was further confirmed by a phenotypic assay, as the katG mutant was more resistant to INH treatment (16 μg∙mL–1 ) than the WT strain[52] (Fig. 5(b)). Because the relative position of the introduced premature stop codon in an open reading frame (ORF) can significantly affect the gene inactivation efficiency, we systematically calculated the possible targetable codons of CTBEengineer in mycobacteria with CRISPR-CBEI [50]. As shown in Fig. 5(c), more than 75%, 60%, and 40% ORFs of the analyzed mycobacterium species (M. tuberculosis, M. smegmatis, and Mycobacterium marinum, respectively) can possibly be targeted by CTBEengineer to introduce at least one premature stop codon within the top 75%, 50%, and 25% of the ORF body, respectively, demonstrating that numerous genes can possibly be inactivated by CTBEengineer in mycobacteria.
《Fig. 5》

Fig. 5. Base editing in M. tuberculosis with CTBEengineer. (a) Bar plots showing the editing efficiencies of CTBEengineer in M. tuberculosis on different sites. Numbering on the bottom indicates the position of the bases in a protospacer, with one being the most PAM-distal base. Arrowheads indicate cytosines with C-to-T conversions. (b) Inactivation of katG by CTBEengineer by generating a premature stop codon. Inactivation of katG makes M. tuberculosis resistant to INH. A box and an inverted box indicate the target sequence and PAM, respectively. (c) Relative position of the earliest induction of stop codons targetable in mycobacteria ORFs (cumulative percentage) by CTBEengineer.
《4. Conclusions》
4. Conclusions
Genetic manipulation is of vital importance in facilitating the study of M. tuberculosis biology and drug-target exploration. Although it is highly desirable, scarless, precise, and markless editing in M. tuberculosis relies on HDR and requires months to years for editing. CRISPR-assisted HDR methods have been developed for rapid and precise genome editing in a number of bacterial species [13,15,17,34]. However, it is not applicable in M. tuberculosis, likely due to the lack of a CRISPR-compatible HDR system. CRISPRi systems using catalytic inactive St1Cas9, SpCas9, or Francisella novicida Cas12a (FnCas12a) have been developed for gene silencing in mycobacteria. However, these systems can only achieve partial gene knockdown and will cause a polar effect in which the operonic genes downstream of the Cas protein binding sites are also silenced [21,22].
To address these challenges in mycobacteria gene editing, we developed highly efficient PAM-expanded St1Cas9 C-to-T and Cto-G base editors for programmed base editing in mycobacteria. These systems can achieve precise single-base substitutions via a single transformation step, thereby substantially reducing the time and efforts required for genetic manipulation. Moreover, the expression of the operonic genes downstream of the Cas protein binding sites that can be silenced by the CRISPRi system [21,22] will not be affected by the base editing systems. In addition, the base editing systems are amenable to highly efficient multiplexed editing—something that is extremely difficult to achieve using serial editing, which is prohibitively time consuming in slowgrowing pathogens. Because only a 20 nt spacer sequence is required for targeting, in addition to being used to perform single-gene editing, the systems can be further engineered into a high-throughput gene knockout screening method. Such a method would allow for the systematic discovery of new drug targets and facilitate new therapeutic method development in mycobacteria.
《Data availability》
Data availability
The plasmids used in this work are available upon request. The whole-genome sequencing data of various mc2155 strains (accession numbers: PRJNA798509) have been deposited at the National Center of Biotechnology Information (NCBI).
《Acknowledgments》
Acknowledgments
We thank Dr. Yi-Cheng Sun for providing the pYC1640 plasmid. This work was supported by the National Natural Science Foundation of China (21922705 (to Quanjiang Ji), 91753127 (to Quanjiang Ji), and 2207783 (to Quanjiang Ji)), the Shanghai Committee of Science and Technology (19QA1406000 (to Quanjiang Ji)), the Emergency Key Program of Guangzhou Laboratory (EKPG21-18 (to Quanjiang Ji)), and General Program of Jiangsu Health Committee Foundation (M2020019 (to Wei Chen)).
《Compliance with ethics guidelines》
Compliance with ethics guidelines
Hongyuan Zhang, Yifei Zhang, Wei-Xiao Wang, Weizhong Chen, Xia Zhang, Xingxu Huang, Wei Chen, and Quanjiang Ji declare that they have no conflict of interest or financial conflicts to disclose.
《Appendix A. Supplementary data》
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.eng.2022.02.013.











 京公网安备 11010502051620号
京公网安备 11010502051620号




