

《1. Introduction》
1. Introduction
Reactions in a homogenous phase are highly efficient for chemical kinetics and are widely accepted in the field of chemical synthesis [1–3]. For example, it is estimated that the reaction rate of homogeneous catalysis is conventionally higher than that of heterogeneous catalysis. Therefore, determining an appropriate solvent to dissolve the reactants is the first step before carrying out a reaction [4–6]. This is because the reactant molecules can easily exhibit high activities in heated solutions or a homogenous phase, in which effective collisions can frequently occur among the fully dispersed reactant molecules [7–9]. Meanwhile, the reactions are constrained by thermodynamic laws; hence, these reactions may be considered practicable only if they are proven to be feasible in terms of thermodynamics.
The aforementioned principles are embodied in copolymer synthesis, particularly for reactions involving the reactants/ monomers presented as solid phase under ambient conditions. Additionally, innumerable chemicals may be employed as the reactants/monomers for copolymerization, and many of them have large molecular weights, which exhibit relatively high activities when dissolved in an appropriate solvent [10,11]. Additionally, endowing homologous copolymers with different properties or with multiple functions has been the trend in current studies [12–14]. Because of the identical reaction type, homologous derivatives can be freely selected, designed, or modified as monomers, as long as their polymerizable functional groups remain unchanged [15,16]. Thus, the polymerization conditions, such as the solvent and temperature, require minor alterations. For example, Hu et al. [17] summarized 21 polymerizable methacrylate derivatives that can be employed to prepare tertiary amine methacrylate-based block copolymers with different pHresponsiveness in water/methanol or tetrahydrofuran (THF). However, this list of available homologous derivatives is not infinite because some desired reactions with specific homologous derivatives may be impracticable in terms of chemical kinetics and thermodynamics.
A desired but impracticable reaction is considered in this study: the copolymerization of 1,3,5-tris(chloromethyl)-2,4,6-trimethyl benzene (TTB) and melamine. With carbonization under specific conditions, the desired copolymer is expected to be used as an ideal precursor for fabricating N-doped porous carbons (NDPCs), which have shown great potential in various applications, such as catalysis, energy storage, electrochemistry, and adsorption [18–21]. The relatively high aromaticity and N content of the polymer precursor are proven to be beneficial for increasing the carbon yield, developed porosity, and N content of the generated NDPCs [22,23]. Therefore, aromatics and N-containing organic compounds have been recognized as ideal monomers to fabricate polymer precursors, such as aryl halides with ursol, ethanediamine, or 3,5- diamino-1,2,4-triazole; among these copolymerizations, Hofmann-type copolymerization can be successfully implemented in specific solvents. These copolymerizations are proven to be so efficient that catalysts are unnecessary [24,25].
At the beginning of this study, it is found that TTB cannot completely react with melamine, as tested in six types of solvents. Using first-principle calculations, it is confirmed that the desired copolymerization is impracticable in terms of thermodynamics and kinetics, either in vacuum or solvent. However, copolymerization is successfully realized using a solvent-free method. This abnormal phenomenon is experimentally confirmed and explained using first-principle calculations and molecular dynamics (MD) simulations, which exhibit completely different scenes between copolymerization with the solvent-free method and that with a solvent. The efforts to implement this ‘‘impracticable” reaction are successful. The as-synthesized copolymer shows the desired potential for producing NDPCs with large surface areas and welldeveloped porosities. The NDPCs exhibit ideal CO2 capture capacities, as well as good selectivity for CO2 and reusability, particularly the NDPC carbonized at 700 °C (6.4 mmol·g–1 at 0 °C and 1 bar, 1 bar = 105 Pa), which can compete with many recently reported NDPCs, such as WSC-500-1, NPC-1-700, and KBM-900 (6.0, 5.1, and 4.4 mmol·g–1 at 0 °C and 1 bar, respectively).
《2. Materials and methods》
2. Materials and methods
《2.1. Materials synthesis》
2.1. Materials synthesis
The copolymerization scheme is shown in Fig. 1. All chemicals were commercially purchased and used as received. Within an N2 atmosphere, TTB (5.0 mmol) and melamine (5.0 mmol) were dissolved in a specific solvent (50 mL) and reflux was maintained for 72 h. In this study, methanol, THF, cyclohexane, and N,N-dimethylformamide (DMF) were chosen as the solvent, and the reaction temperature was set as 65, 70, 80, and 155 °C, respectively. Afterward, dimethylsulfoxide (DMSO) and glycol were selected as the solvents and charged with N2 up to 0.5 MPa, and the reaction was carried out in an autoclave for 72 h at 210 °C, which is the temperature of the solvent-free method.
《Fig. 1》
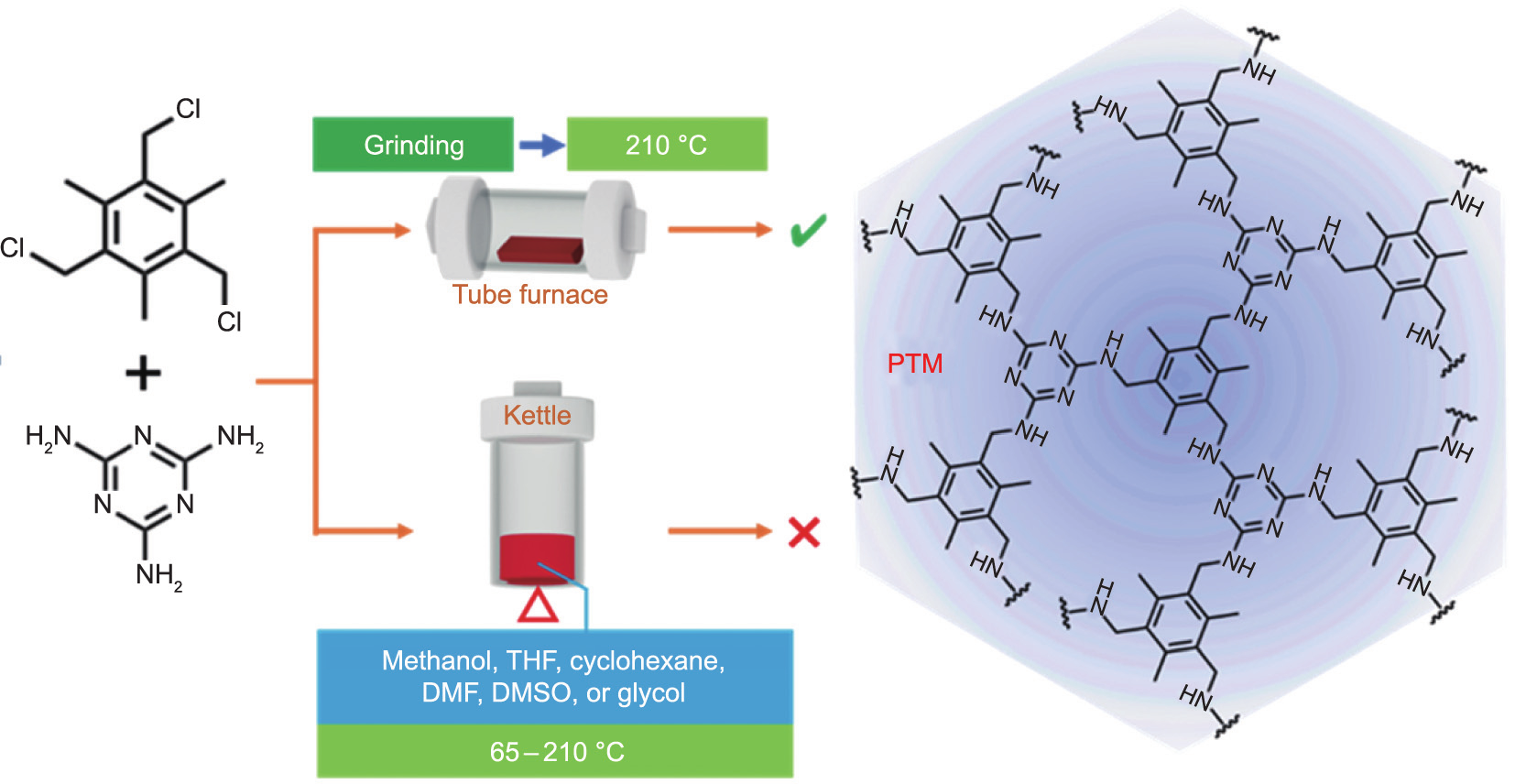
Fig. 1. The scheme of PTM synthesized in the solvent or with solvent-free method.
For the solvent-free method, TTB (5.0 mmol) and melamine (5.0 mmol) were ground for 15 min in an agate mortar; NaCl powder (0.06 g) was used as the grinding aid [26–28]. With N2 protection, the mixture was transferred into a tube furnace and maintained at 210 °C for 24 h to obtain the brown copolymer powder. Afterward, the copolymer powder was purified in boiling methanol for 3 h to remove the unreacted monomers, followed by successive elution with methanol, CH2Cl2, and deionized water to further remove the residual monomers and NaCl. After vacuum drying at 100 °C for 24 h, the copolymer, polymerized TTB and melamine, code-named PTM, was finally obtained.
The PTM-generated NDPC, code-named PTMC-x, where x indicates the carbonization temperature, was prepared as follows. The PTM was dispersed in an ethanol/water (1/1, v/v) solution containing KOH (30 wt%). After sufficient impregnation and evaporation, the mixture with N2 protection was carbonized at the prescribed temperatures (500, 600, 700, and 800 °C) for 60 min. PTMC-x was finally obtained after treating with 2 mol·L–1 HCl to remove the residual KOH, washing with deionized water, and drying in a vacuum at 120 °C for 24 h.
《2.2. Characterization》
2.2. Characterization
A Nicolet Nexus 470 spectrometer with a KBr wafer was used to record the Fourier-transform infrared (FTIR, Nicolet iS10, USA) spectra. The solid-state 13C nuclear magnetic resonance (13C NMR) spectrum was recorded using an Agilent-NMR-vnmrs600 spectrometer (USA). A Bruker D8 Advance diffractometer was employed to record the X-ray powder diffraction (XRPD, Bruker D8 Advance, Germany) patterns with Cu Kα radiation at 40 kV and 40 mA. Elemental analysis was carried out using an Elementar Vario EL elemental analyzer (Germany). Scanning electron microscopy (SEM) images were obtained using a Hitachi S-4800 and FEI Tecnai G2T20 electron microscope (Japan). Transmission electron microscopy (TEM) images of the materials were captured using a JEM-2010 UHR electron microscope operated at 200 kV (Japan). Raman spectra were recorded using an HR800 UV Raman microspectrometer (HORIBA, USA). X-ray photoelectron spectroscopy (XPS) was performed using an ESCALAB-220I-XL device (VG Scientific, UK). After degassing at 150 °C for 4 h, the N2 adsorption–desorption isotherms of the samples were measured at 77 K using a Micromeritics ASAP 2020 analyzer (Micromeritics, USA). The Brunauer–Emmett–Teller (BET) surface areas were calculated using data from the adsorption branch. The total pore volumes were derived from uptake at a relative pressure of 0.95. The pore size distribution was estimated using non-local densityfunctional theory (NLDFT). Static adsorption experiments of CO2 (99.999%) and N2 (99.999%) over the adsorbents at 0 and 25 °C were performed using an ASAP 2020 analyzer. The free space was determined using helium (99.999%), assuming that helium was not adsorbed at the tested temperatures.
《3. Results and discussion》
3. Results and discussion
《3.1. Mechanism of the copolymerization》
3.1. Mechanism of the copolymerization
Although it has been proven that Hofmann-type copolymerization was sufficiently efficient, it can be experimentally confirmed that, with any of the six representative solvents, namely methanol, THF, cyclohexane, DMF, DMSO, and glycol, no copolymer products were generated. This implies that the reaction between TTB and melamine was forbidden in terms of thermodynamics or kinetics, as estimated using the first-principle calculations; the computational details are given in the Appendix A [29–32]. As can be seen in Fig. 2(a), the reaction between TTB and melamine occurs in one step only, which exhibits a typical SN2 reaction model. The aromatic ring of TTB was parallel to the azacyclo-ring of melamine, which was the orientation of the effective collision, such that one of the amino groups of melamine suppressed the –Cl group of TTB, and the Cl atom was stripped. Meanwhile, a transition state (TS) was generated. The activated Cl atom instantly collected an H atom of the suppressed amino group, and thus, a –CH2–NH– bond was formed with a molecule of HCl. The calculated intrinsic free energy change (298 K) of this reaction in vacuum was 4.5 kcal·mol–1 (1 kcal = 4.18 kJ), which indicated that the desired reaction was forbidden in terms of thermodynamics at ambient temperature. Meanwhile, the calculated reaction energy barrier in a vacuum reached 37.2 kcal·mol–1 , which was sufficiently high to inhibit the reaction unless the temperature was appropriately increased.
《Fig. 2》
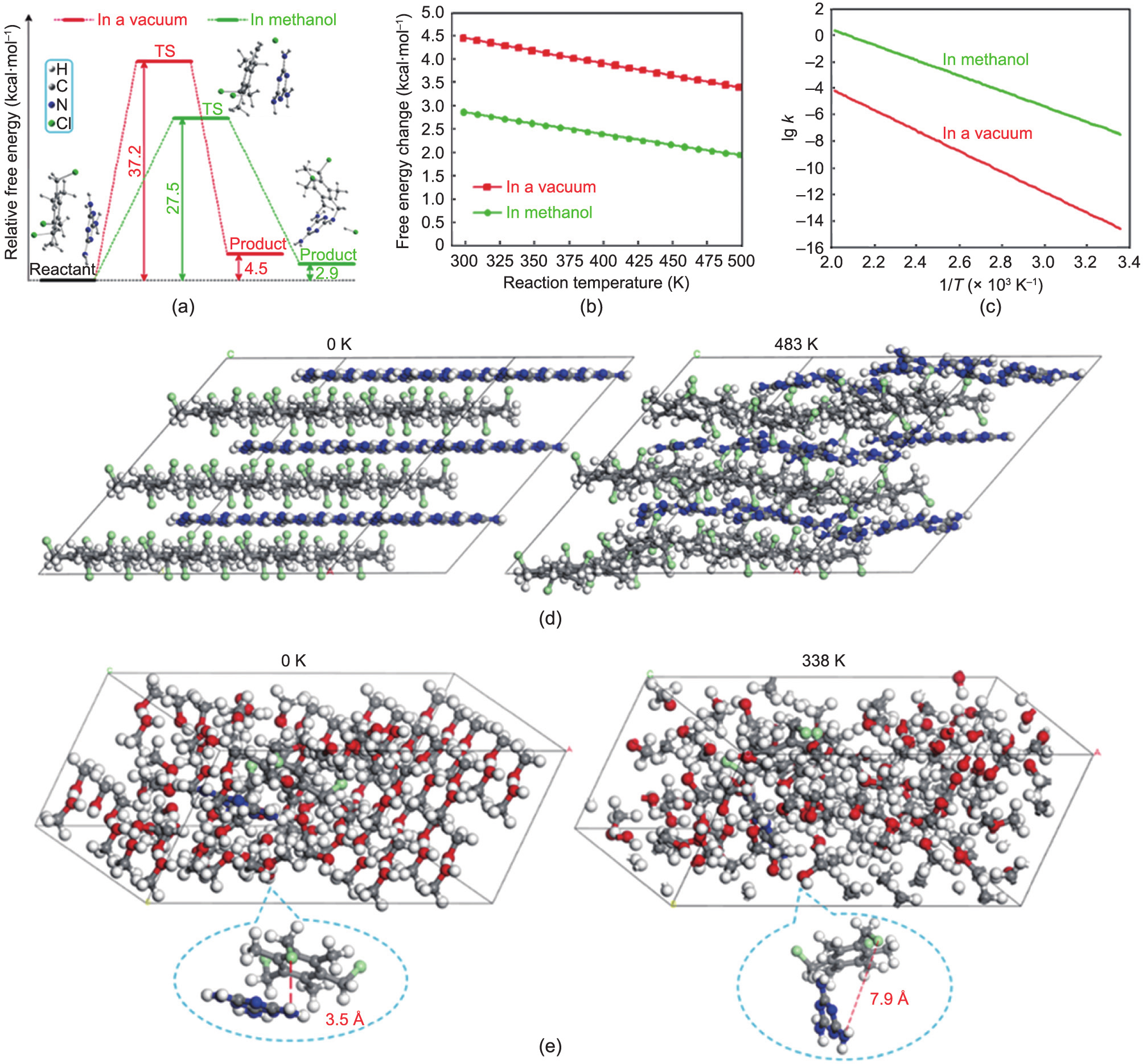
Fig. 2. (a) First-principle calculated potential energy profiles of the copolymerization in a vacuum and with the reaction field of methanol, (b) free energy changes of the copolymerization with the variational temperature, (c) the relationship between the logarithm of reaction rate constants and the reciprocal of temperature, (d) the relative displacements of the reactant molecules impelled at 483 K for 50 ps compared to that at 0 K for the solvent-free case simulated with MD, and (e) the relative displacements of a pair of reactant molecules surrounded by 100 methanol molecules impelled at 338 K for 50 ps compared to that at 0 K simulated with MD.
Using the solvent, a self-consistent reaction field based on the polarizable continuum model was employed to simulate the solvent molecules in the vicinity. Methanol was used in this study. The calculation results showed that the relative position between TTB and melamine could not significantly change if the reaction occurred, even with methanol. As can be seen in Fig. 2(a), both the free energy change of the reaction (2.9 kcal·mol–1 ) and the energy barrier (27.5 kcal·mol–1 ) are reduced because of the methanol. However, the reaction was still forbidden at 298 K. Furthermore, using the Gibbs–Helmholtz equation, the free energy changes of the reaction in vacuum and that in methanol were calculated at temperatures ranging from 298 to 498 K, covering each operation temperature of the copolymerization in this study (Fig. 2 (b)). It was observed that the free energy change at all the investigated temperatures remained positive, explaining the infeasibility of the reaction in all investigated solvents and at relatively high temperatures. The impact of temperature on the reaction rate constant k was calculated, as shown in Fig. 2(c). As the temperature increased, the k value rapidly increased. For example, the k value of the reaction in methanol was increased from 3.89 × 10–8 s–1 at 298 K to 7.60 × 10–6 s–1 at 338 K. When the temperature was increased to 483 K, the k value of the reaction reached 2.25 × 10–5 s–1 in a vacuum, which was the theoretical k value of the solvent-free method at 210 °C. The high increase in k value indicated that the reaction may occur easily in terms of chemical kinetics, whereas the positive free-energy change indicated that the reaction cannot occur spontaneously unless the solvent-free method breaks through such an equilibrium. Notably, HCl was the by-product of the copolymerization, and the operation temperature of the DMSO or glycol was the same as that of the solvent-free method. In contrast to the reaction in the solvents, the solvent-free method immediately released the generated HCl into the atmosphere, such that the complex structure of the product was dissociated, and thus, the reaction between TTB and melamine was pushed in the right direction.
The MD simulations indicated another important reason why the reaction occurred only with the solvent-free method: the solid phase. The MD simulation parameters of the unit cell of the TTB and melamine (1:1) mixture were consistent with the results of the first-principle calculations, which indicates the accuracy and reliability of the MD simulation (Table S1 in Appendix A). As can be seen in Fig. 2(d), the MD simulation for 27 TTB and 27 melamine molecules at 483 K for 50 ps describes the thermal motions of the system under the experimental temperature, which is sufficiently high to activate the reaction but slightly changes the relative positions of the monomer molecules in the solid phase. Therefore, the TTB and melamine molecules can impact each other in parallel and initiate the reaction. However, the monomer molecules dispersed among the methanol molecules were significantly disturbed. As can be seen in Fig. 2(e), with a pair of TTB and melamine molecules coexisting with 100 methanol molecules, the MD simulations at 338 K for 50 ps show that the TTB and melamine molecules are separated by the methanol molecules. Further, the distance between the –NH2 and –Cl groups was stretched from 3.5 Å at 0 K to 7.9 Å at 338 K, which was too long to initiate the reaction. The importance of effective distance between the monomer molecules was proven by another noticeable experimental fact. No copolymer product was generated, even with the solvent-free method, if the temperature was increased above 220 °C, under which the TTB in the mixture started melting and the monomer molecules exhibited increased activities. However, the excessively activated TTB molecules in the melting phase were separated from the melamine molecules, as in the solvent.
The aforementioned theoretical and experimental results reveal two underlying reasons for the successful copolymerization of TTB and melamine using the solvent-free method: The reaction equilibrium was pushed by the timely released by-product, HCl, and the thermal motions of the activated monomer molecules were confined in the solid phase. Additionally, the thermal condensation of melamine played a role during the solvent-free treatment to realize copolymerization, ensuring the stability of the final NDPC products.
《3.2. Characterization of the copolymer》
3.2. Characterization of the copolymer
The successful fabrication of PTM was confirmed by the FTIR spectrum shown in Fig. 3(a). The bands located at 3348 and 3216 cm–1 were attributed to the stretching vibration of –NH–, and that located at 2916 cm–1 was caused by the stretching vibration of alkyl C–H. The strong bands at 1682 and 1643 cm–1 indicated the presence of aromatic rings, which were hidden by the strongest band at 1547 cm–1 caused by the in-plane bending vibration of –NH–. The peaks at 1458 and 1335 cm–1 were attributed to the in-plane bending vibrations of the –CH2– groups that were generated during the copolymerization and the –CH3 groups attached to the aromatic rings, respectively. In the fingerprint region, the two adsorption bands at 1169 and 1022 cm–1 were caused by the stretching vibration of aliphatic C–N bonds formed during the copolymerization. The two weak peaks located at 810 and 780 cm–1 were ascribed to the presence of C–N bonds in the melamine moieties, and the vibration of –NH– afforded a peak at 690 cm–1 . The 13C NMR spectrum provided more evidence for the formation of the copolymer. As can be seen in Fig. 3(b), the peaks located at 38.6 and 72.3 ppm indicate the presence of the C atoms in the newly formed –CH2–NH– linkages, and that located at 13.2 ppm corresponds to the C atoms of the –CH3 groups. The two peaks at 130.8 and 162.9 ppm were caused by the two types of aromatic C atoms, and that located at 187.7 ppm was ascribed to the C atoms of the triazine rings. According to the FTIR and 13C NMR spectrum results, the reaction between TTB and melamine was mainly a single substitution reaction, namely the formation of – CH2–NH– linkages. This was because of the absence of catalysts and the use of a strict molar ratio of the reactants based on the theoretical structure of the as-synthesized polymer. This approach has been proven to be reliable in previous reports [23,24,33].
《Fig. 3》
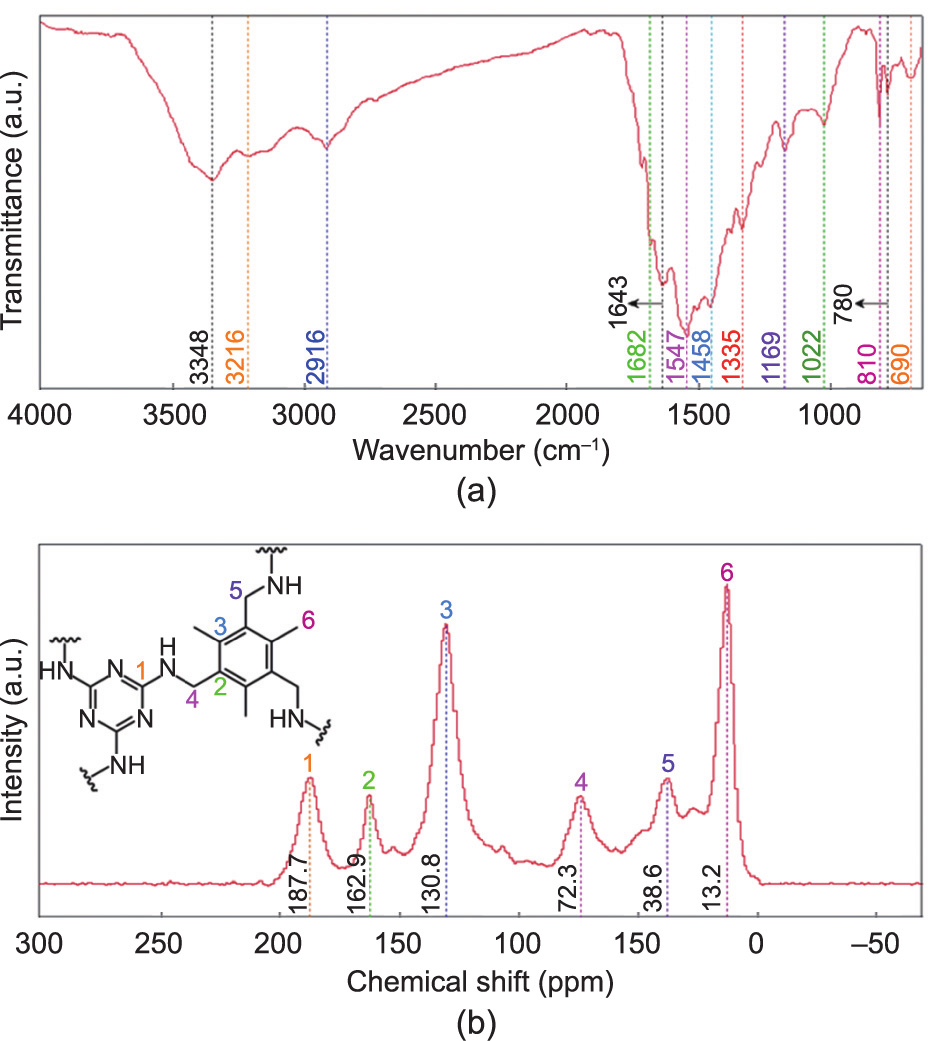
Fig. 3. (a) FTIR spectrum and (b) 13C NMR spectrum of PTM. a.u.: arbitrary unit.
The successful fabrication of the PTMs was supported by elemental analysis. As can be seen in Table 1, the experimental elemental contents of C, H, and N are 63.9 wt %, 6.6 wt %, and 27.5 wt %, respectively, which are consistent with the theoretical values (C, 63.8%; H, 6.4%; N, 29.8%). Normalized by differences, other elemental contents were mainly attributed to the presence of O and trace Cl from the monomer of TTB. Because of the rotatable or flexible σ-type –CH2–NH– linkages, the as-synthesized PTM was confirmed to be amorphous; this is because the macromolecules of the PTM could fold and curl randomly during the polymer chain growth. As can be seen in Figs. S1 and S2 in Appendix A, a typical amorphous state of the PTM is exhibited by the XRPD pattern and SEM image. The morphology of the PTM at the molecular level was simulated using first-principle calculations for a molecular fragment of the PTM copolymerized by 7 TTB and 15 melamine molecules (Fig. S3 in Appendix A). The PTM molecular fragment folded and curled to the extent possible to shrink; this was because of the rotatable σ-type –CH2–NH– linkages and the strong intramolecular van der Waals forces. The folded and curled configuration of the PTM, the desired precursor of the NDPCs, resulted in short distances between the aromatic and triazine rings, and the PTM easily self-condensed once the radicals or other reactive intermediates were formed. Meanwhile, the N atoms were easily doped into the carbon crystallites to form NDPCs with ideal textural properties and application performances.
《Table 1》
Table 1 Textural parameters and elemental analysis of PTM and PTMCs.
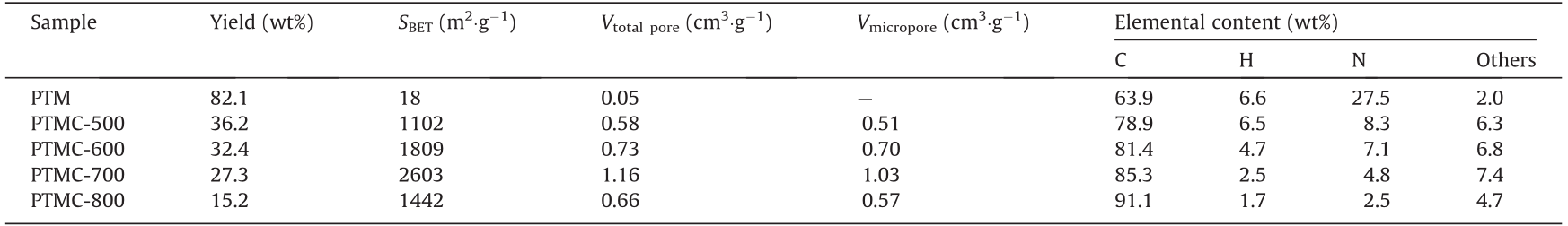
SBET: The BET specific surface area.
《3.3. PTM-generated NDPCs》
3.3. PTM-generated NDPCs
By carbonization, a series of NDPCs, code-named PTMCs, were derived from PTM. As can be seen in Fig. S4 in Appendix A, as the carbonization temperature increased, the characteristic bands of the PTM on the FTIR spectra gradually disappeared, indicating the development of carbon crystallites with the successive dissociation of the functional groups. Further, the evolution of the carbonaceous textural structures was demonstrated by Raman spectroscopy. As can be seen in Fig. S5 in Appendix A, two characteristic peaks centered at 1345 and 1581 cm–1 are observed, corresponding to the D-band representing the disordered structure or the carbonaceous crystallite defects and the G-band indicating the in-plane vibrations of the sp2 carbon atoms, respectively. As the carbonization temperature increased from 500 to 800 °C, the intensity ratio of the D-/G-band continuously decreased, indicating the enhanced graphitization of the NDPCs. According to the XRPD patterns (Fig. S6 in Appendix A), PTMCs present the typical features of amorphous NDPCs without apparent sharp diffraction peaks. The two dispersive diffraction peaks located at approximately 25° and 43° were ascribed to the reflection of the (002) and (100) graphitic planes of the NDPCs, respectively. As the carbonization temperature increased, the two dispersive diffraction peaks stood out from the background, indicating the developed graphitization of the NDPCs, which is consistent with the Raman spectra. As can be seen in Table 1, the PTM exhibits good potential for the fabrication of the NDPCs with satisfactory yields; for instance, to the best of our knowledge, 27.3 wt% for PTMC-700 was seldom reached with other artificial polymer precursors. According to the XPS results (Fig. S7 in Appendix A), three N-doped patterns were detected on the NDPCs: pyrrolic N (N-5), pyridinic N (N-6), and quaternary N (N-Q) [24]. When the carbonization temperature was increased from 500 to 800 °C, the N content was a trade-off for the graphitization of the NDPCs, whereas N-5 sites were more stable than the other two N-doped sites; thus, more N-5 sites remained. The total N content of the PTMCs, for example, 4.8 wt % for PTMC-700, can compete with those of some representative NDPCs in other reports (Fig. S5 in Appendix A).
The most remarkable advantage of the PTM, as the precursor of the NDPCs, was reflected in the textural properties of the PTMCs, which were investigated using the N2 adsorption–desorption isotherms; the calculated textural parameters are shown in Table 1. As can be seen in Fig. S8(a) in Appendix A, all the PTMCs feature typical type-I isotherms with rapid N2 adsorption in the low P/P0 range, indicating the presence of abundant micropores. The hysteresis loops of the isotherms for all PTMCs were not obvious, which indicated that the few mesoporous structures observed, if any, were caused by particle stacking. The PTM had few pores, and its BET specific surface area was considerably low because, as the precursor of the NDPCs, the PTM was amorphous and pyknotic, as mentioned earlier. However, the BET specific surface areas of the PTMCs were sufficiently high after carbonization. In contrast to other PTMCs (1102, 1809, and 1442 m2 ·g–1 for PTMC-500, PTMC 600, and PTMC-800, respectively), PTMC-700 presented the highest BET specific surface area of 2603 m2 ·g–1 , which was higher than those of many other reported NDPCs (Table S2). The reduced BET specific surface area of PTMC-800 was attributed to excessive carbonization, which caused the further disintegration of the carbon crystallites. The variation tendency of the BET specific surface area was consistent with that of the porosity of the PTMCs. As can be seen in Fig. S8(b) in Appendix A and Table 1, the pore volume of the PTMC increased until the carbonization temperature reached 700 °C (1.16 cm3 ·g–1 for PTMC-700); the micropore volume also increased (1.03 cm3 ·g–1 for PTMC-700). For each PTMC, more than 85% of the pore volume was contributed by micropores. Additionally, the changes in the porosity of the PTMCs were observed in the SEM images (Fig. S9 in Appendix A). The temperature of 500 °C was proven to be sufficient to carbonize the PTM, as shown in the FTIR spectra, whereas the pore structures of PTMC-500 were undeveloped. As the carbonization temperature increased to 600 °C and then to 700 °C, the porosity of the PTMCs was developed stepwise, and this was shown in the cross-section of PTMC700. The excessive carbonization at 800 °C caused the ablation of the carbon texture of PTMC-800, and the BET specific surface area and pore volume decreased at this point Fig. S8(b) in Appendix A.
A large BET specific surface area and well-developed porosity usually indicate the good potential of the material for many applications, such as gas storage and separation. Thus, the PTMCs were used as adsorbents to capture CO2 in this study. As shown in Fig. 4, compared with other PTMC counterparts, PTMC-700 exhibits higher CO2 capture capacities of 6.4 mmol·g–1 (at 0 °C, 1 bar) and 3.4 mmol·g–1 (at 25 °C, 1 bar) because it possesses the largest BET surface area and pore volume, particularly the micropore volume, which was proven to be crucial for the NDPCs to confine CO2 molecules. Additionally, as shown in Table S2, PTMC-700 not only exhibits higher BET specific surface area but also competitive CO2 capture capacity compared with other reported representative NDPCs [34–45], such as WSC-500-1, NPC-1-700, and KBM-900 (6.0, 5.1, and 4.4 mmol·g–1 at 0 °C and 1 bar, respectively). The good selectivity for CO2 toward N2 over PTMCs was confirmed by the ideal adsorption solution theory (Figs. S10 and S11 in Appendix A). The CO2/N2 (85/15, v/v) selectivity values of PTMC-700 reached 18 and 32 at 0 and 25 °C, respectively, which are comparable with those of some other representative NDPCs (Table S2). This was largely due to the presence of sufficient N-doped sites that acted as affiliative sites for CO2 molecules. Notably, the residual N content of the NDPCs was low because of carbonization. In this study, however, the N content of PTMC-700 reached 4.8 wt%, which was higher than those reported for other representative NDPCs (Table S2).
《Fig. 4》

Fig. 4. CO2 adsorption isotherms of PTMCs at 0 and 25 °C.
The CO2 adsorption performance of the NDPCs was mainly determined by the synergetic effect of three factors: micropores, N-doped sites, and surface areas [23]. The micropores afforded the microenvironments for confining CO2 molecules, and the N species doped in the NDPCs were beneficial for CO2 anchoring because the N species could create a strong van der Waals force between the adsorbent surface and CO2 molecules [46] and successively enhance the CO2 uptake and selectivity. In this study, the pore texture of the NDPC dominated the contribution to the CO2 capture capacity because of the congruent relationship with the BET specific surface areas, whereas a considerable contribution from the N-doped sites was observed, allowing for a higher CO2/ N2 selectivity of PTMC-500 than that of PTMC-800 (Fig. S11 in Appendix A).
Further, the physical adsorption between the CO2 molecules and NDPCs was verified using the calculated isosteric heats of adsorption (Qst). All the Qst values were lower than 30 kJ·mol–1 , based on the CO2 isotherm data at temperatures of 0 and 25 °C (Fig. S12 in Appendix A). Reusability is another criterion for evaluating adsorbents. To examine the reusability, PTMC-700 was evacuated at 130 C for 100 min for regeneration, and then saturated with CO2 up to 1 bar at 0 °C. As can be seen in Fig. S13 in Appendix A, even after six CO2 adsorption–desorption cycles, the attenuation of the CO2 capture capacity of PTMC-700 was not observed. There after, the N2 adsorption–desorption test and the pore distribution analysis for PTMC-700 after the reusability test were performed to investigate the changes in the textural properties (Fig. S14 in Appendix A). It was found that the BET specific surface area (2600 m2 ·g–1 ), the total pore volume (1.16 cm3 ·g–1 ), and the pore size distribution of the PTMC-700 were well maintained. This further confirmed the good reusability of PTMC-700 for CO2 adsorption. The satisfactory yields of the NDPCs, the competitive N contents and textural properties, the good CO2 capture capacity and selectivity, and the perfect reusability collectively demonstrated that the PTMCs generated from the PTM were in line with expectations. Additionally, the PTM prepared only through the solvent-free method was a satisfactory precursor for the NDPCs.
As for the synthetic methodology, one of the advantages of this work is that it provides a potential approach to realize those Hofmann alkylation reactions that were ‘‘impracticable” in solvents. Additionally, according to previous reports, some target materials based on the Hofmann alkylation reaction had to be synthesized with a corrosive strong alkaline catalyst in the solvent [47], and the purification of the product was complex. Therefore, another advantage of this work is that, with the solvent-free approach, corrosive catalysts are unnecessary, and the purification of the product is considerably easier than that with solvents. Additionally, with the approach proposed in this study, some copolymerization reactions can be implemented to fabricate polymer precursors with specific molecular structures beneficial to the formation of the pore textures of porous carbons and with specific to-begrafted groups that can be subsequently substituted by a specified content of N-containing groups, such as amino groups [34]. In theory, it is possible to independently regulate the pore texture and Nfunctional group concentration of the subsequent porous carbon products.
《4. Conclusions》
4. Conclusions
In summary, the desired copolymerization of TTB and melamine, which cannot take place in solvents, was successfully achieved using a solvent-free method. This reaction was proven to be impracticable in terms of chemical kinetics and thermodynamics using first-principle calculations. However, the immediate release of the by-product, HCl, and the sufficiently high reaction temperature in the solvent-free method broke through the inhibition of thermodynamics and kinetics, respectively, which cannot be realized in solvent environments. Moreover, MD simulations demonstrated that an impacted complex structure of reactant molecules, which can be well maintained in the solid phase with a solvent-free method, is crucial for implementing the reaction. However, the solvent molecules separate the reactant molecules, thereby inhibiting the effective collisions between the reactant molecules. Efforts to fabricate such desired copolymers with high aromaticity and high N content were proven to be successful. A series of NDPCs possessing satisfactory carbon yields, high N contents, well-developed porosities, and sufficiently high BET specific surface areas were generated through the carbonization of the copolymer. The NDPCs exhibited high CO2 capture capacities, good selectivity, and ideal reusability, particularly those carbonized at 700 °C, because of the perfect synergetic effect of the pore texture and N-doped sites. These features make the NDPCs competitive with those in other reports, and the underlying mechanisms of the copolymerization exhibited in this study might stimulate the review of some other ‘‘impracticable” but desired reactions.
《Acknowledgments》
Acknowledgments
We acknowledge the financial support for this work from the National Natural Science Foundation of China (22178163, 21808105, 21722606, 21878149, and 22078155), the Natural Science Foundation of Jiangsu Province (BK20180709), and the National Science Fund for Distinguished Young Scholars (22125804). We are also grateful to the High Performance Computing Center of Nanjing Tech University for providing the computational resources.
《Compliance with ethics guidelines》
Compliance with ethics guidelines
Shi-Chao Qi, Xiao-Jie Lu, Yin-Cong Lou, Rui Zhou, Ding-Ming Xue, Xiao-Qin Liu, and Lin-Bing Sun declare that they have no financial or other conflicts of interest to disclose.
《Appendix A. Supplementary data》
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.eng.2021.07.031.











 京公网安备 11010502051620号
京公网安备 11010502051620号




