

《1. Introduction》
1. Introduction
In biological and chemical studies, the traditional biochemical bulk assay has been widely used to uncover the molecular properties, functions, and mechanisms of biomacromolecules, including DNA, RNA, and protein [1–4]. However, the bulk assay only provides an average value, as it ignores individual differences between molecules. To comprehensively understand molecular properties and mechanisms, single-molecule methods have emerged. These methods are powerful tools for analyzing and studying molecules in highly heterogeneous systems. Single-molecule methods generally include single-molecule detection methods and singlemolecule manipulation methods. In detail, single-molecule detection techniques (i.e., single-molecule fluorescence [5], nanopores [6], single-molecule super-resolution (SM-SR) microscopy [7], etc.) can be used to observe and track in real time the rapid transition process between different states of a biomolecule during fluorescence changes; while single-molecule manipulation techniques (i.e., magnetic tweezers [8], optical tweezers [9], and atomic force microscopy (AFM) [10]) can be used to directly manipulate a molecule in order to detect the dynamic processes of the molecule under tension. The applied force on the biomolecule can decrease the transition energy barrier, which accelerates the conformation transition rate and shortens the detection timescale. Another benefit of single-molecule manipulation is that these methods can mimic the forces generated in vivo, thereby determining and quantifying the effects of forces on physiological and pathological processes [11,12]. Hence, single-molecule methods reveal the precise dynamics of biochemical or biophysical reactions by analyzing the kinetic and thermodynamic information of a single biomacromolecule such as DNA.
《Fig. 1》
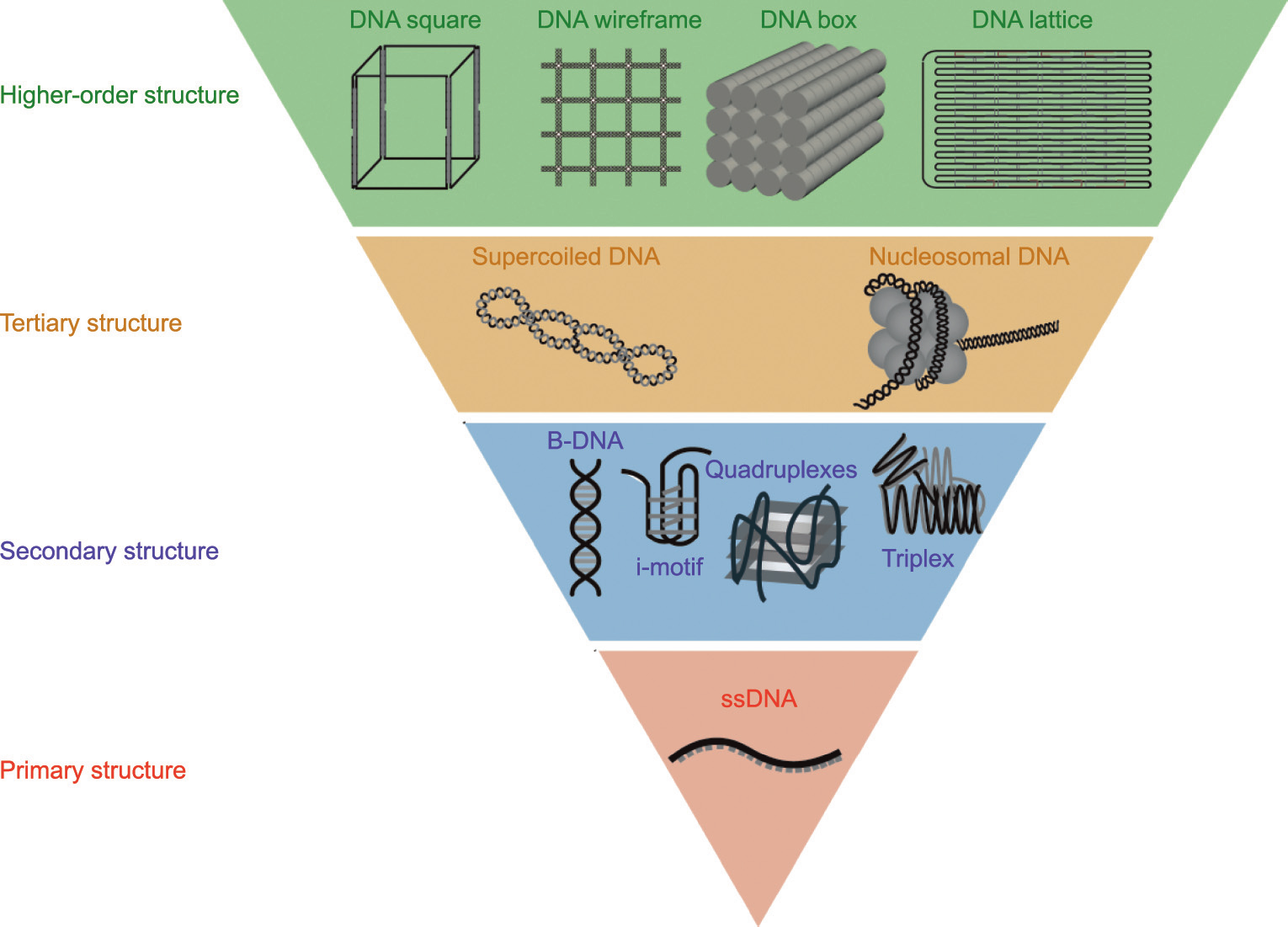
Fig. 1. The DNA primary structure and different higher-order structures. The DNA primary structure refers to the single-stranded DNA (ssDNA). Based on the inter- and intrastrand interaction, DNA can also form complex spatial structures and topologies, which are secondary structures and tertiary structures. The high-order structure refers to DNA artificial nanostructures. B-DNA: B-form DNA.
As a fundamental biomacromolecule in vivo, DNA plays a vital role in the storage of genetic information. Based on the interactions between base pairs, strands of DNA form the special spatial structure of DNA molecules that affects the function of DNA. These DNA spatial structures are commonly classified by the complexity of the DNA structure (Fig. 1). According to the Watson–Crick base-pair principle, a single-stranded DNA (ssDNA) oligonucleotide (primary structure) forms a double-stranded DNA (dsDNA) helix due to interactions between A–T and C–G in vivo [13]. A DNA strand can also form more special spatial structures—such as the Gquadruplex (G4) [14], the i-motif [15], three- or four-way junctions [16,17], and the triplex [18]—via inter- or intra-strand interactions [19]. These structures are known as DNA secondary structures. DNA can also form tertiary structures, such as supercoiled DNA [20] and nucleosomal DNA [21]. Aside from these biological function-related structures, DNA can form artificial higher-order nanostructures, such as DNA origami and DNA bricks [22–25]. The DNA structures mentioned above have various chemical and physical properties. Detecting the properties of DNA structures is important in understanding the biological or chemical functions of DNA.
To date, many methods have been developed for detecting different DNA structures. Based on the optical activities of DNA, ultraviolet (UV)-visible spectroscopy [26–28] and circular dichroism (CD) spectroscopy [29–31] have been used to detect DNA conformation changes and interactions with ligands [32]. In addition, nuclear magnetic resonance (NMR), X-ray diffraction, and cryoelectron microscopy (cryo-EM) techniques are frequently used to determine the static or dynamic structure of DNA [29,33,34]. To detect larger scale static DNA nanostructures, transmission electron microscopy (TEM) and AFM are applied to observe shapes and morphologies. During the past two decades, single-molecule methods have provided new insights into measurements of the chemical and physical properties of DNA structures.
In this review, we classify DNA structures into primary, secondary, tertiary, and higher-order structures. We then outline the basic principles of single-molecule techniques and their application to characterizing different DNA structures. Finally, we discuss the advantages of single-molecule techniques and their future development.
The primary structure of DNA is ssDNA. Based on inter- and intra-strand interactions, DNA can also form complex spatial structures and topologies, known as secondary and tertiary structures. The term ‘‘higher-order DNA structures” refers herein to artificial DNA nanostructures.
《2. Single-molecule techniques》
2. Single-molecule techniques
《2.1. Single-molecule manipulation techniques》
2.1. Single-molecule manipulation techniques
With the continuous development of single-molecule manipulation techniques, these techniques continue to increase in number. At present they include the biomembrane force probe [35], flow-induced stretching [36,37], microneedle manipulation [38], AFM, optical tweezers, and magnetic tweezers. The last three methods are the most commonly used, and are the focus of this review (Table 1).
《Table 1》
Table 1 Comparison of single-molecule manipulation techniques.

2.1.1. Atomic force microscopy–single-molecule force spectroscopy (AFM–SMFS)
AFM–SMFS can be used to manipulate individual molecules and measure intramolecular interaction forces (Fig. 2(a)). In this technique, the surface is modified with substrate molecules, while target molecules that can bind to the substrate molecules are modified on the tip of the AFM–SMFS cantilever. Moving the tip so that it can bind to the substrate molecules and then shrinking the tip at a constant speed will cause deflection of the cantilever. The elasticity of the cantilever obeys Hooke’s law, so the force applied on the tethered molecule can be calculated by the deflection and the spring constant of the cantilever. From this, the force-extension curve (FEC) can be obtained.
《Fig. 2》

Fig. 2. Schematic overview of single-molecule manipulation used to detect DNA structures. (a) AFM–SMFS; (b) magnetic tweezers; (c) optical tweezers.
2.1.2. Magnetic tweezers
The basic principle of magnetic tweezers is that magnetic particles placed in a magnetic field gradient experience the same force as the magnetic field gradient. In general, two magnets must be used together, generating tension and torque. This pair of magnets is suspended above the sample flow channel, which exposes the magnetic field below. Inside the flow channel, biomacromolecules are connected between the bottom of the flow channel and the small magnetic beads. The magnetic beads are manipulated by an external magnetic field, thereby controlling the target molecules attached to the beads. Below the flow channel, there is a microscope objective connected to a charge-coupled device (CCD) camera that transfers the observed image to the CCD camera; then, the CCD converts the image into an electrical signal and transmits it to the computer. When a parallel beam of light hits the magnetic beads, light scattering occurs. The scattered light interferes with the unscattered light, causing concentric circles to form around the image captured by the camera (Fig. 2(b)). Magnetic tweezers have several advantages, such as a strong force and ease of operation; moreover, the force provided by magnetic tweezers can be adjusted in the range of 0.1–100.0 pN [39].
2.1.3. Optical tweezers
A focused beam of light acting on objects with an index of refraction higher than the index of refraction of the surrounding medium can generate an optical gradient force. This principle was discovered by Ashkin in 1970 [40]—a discovery that promoted the emergence and maturity of optical tweezers technology [41]. Optical tweezers use a beam of light to capture particles (Fig. 2(c)); and the light must produce the lowest point of potential and form an optical trap. When the barrier of the optical trap is greater than the kinetic energy of an object, the object will be stably bound in the optical trap. Light refracts when it hits the particle. Due to the change in photon momentum, the particle is subject to a reaction force. The resultant force of multiple light rays binds the particle in the center. At this time, the particle can be moved by moving the light field, to realize the function of manipulating the beads like tweezers. Biological macromolecules can also be manipulated by connecting them to particles captured in optical traps. The position of the directional moving beam can control the distance between the two ends of the biomacromolecule, so it can accurately apply the same force to the biomacromolecule as the particle movement direction. Particle sizes ranging from about 20 nm to a few microns can be stably captured [42–45].
《2.2. Single-molecule detection techniques》
2.2. Single-molecule detection techniques
Single-molecule detection techniques can be applied to determine the dynamic characteristics and interactions of biomolecules by detecting and imaging individual molecules in solution and recording the behavior of single molecules in real time [46].
2.2.1. AFM imaging
Another major application of AFM is imaging. When AFM is used as an imaging tool, the basic working principle is that the tip approaches the sample until interaction forces between the tip and the sample cause deformation of the cantilever. The surface profile of the sample can then be reconstructed by recording the deformation of the cantilever. When scanning the sample, the user can measure the vertical position of the tip by means of a laser reflected from the cantilever to the position-sensitive photodetector. AFM imaging resolution can reach atomic levels: up to 2.00 nm for lateral resolution and 0.01 nm for longitudinal resolution.
2.2.2. Single-molecule fluorescence techniques
The basic principle of single-molecule fluorescence is to detect the light emitted by a fluorophore attached to the studied molecule. The fluorescent group in the ground state absorbs light from an external light source, is excited, and emits fluorescent light in 1 × 10–9 –1 × 10–7 s [47]. In single-molecule fluorescence, fluorescent molecules can be attached to DNA, detected, and monitored within a short time [48]. Using single-molecule fluorescence techniques, it is possible to characterize the different conformational states of higher-order DNA structures [49]. Single-molecule fluorescence techniques are widely used for probing different conformations of secondary DNA structures.
In single-molecule fluorescence resonance energy transfer (smFRET) techniques, two differently colored dyes are positioned at specific locations on the host molecule (Fig. 3(a)). Fluorescence resonance energy transfer (FRET) is based on a quantum interaction mechanism between two fluorescent dyes that are close to each other. When the distance between the donor and the acceptor is close enough and the donor is excited by an external light source, the acceptor emits part of the light through a nonradiative resonance energy transfer mechanism between the donor and the acceptor. The efficiency of the energy transfer depends on the distance between the donor and the acceptor:

where R0 is the donor–acceptor distance with a FRET efficiency of 50%, and r is the donor–acceptor distance. The research field of smFRET is expanding, and smFRET can be used in enzymatic reactions and molecular folding and conformational transitions.
2.2.3. Nanopore technology
As a novel platform for single-molecule detection, nanopore technology can distinguish the differences between adenine (A), thymine (T), cytosine (C), guanine (G), and other mononucleoside bases. Nanopore technology has the advantages of high resolution and high throughput. An insulating membrane separates the electrolyte chambers into cis and trans chambers, with one nanosized pore connecting the two chambers (Fig. 3(b)). When an electric field is applied at two ends of a nanopore, occupation of the nanoscale pore by the target molecule leads to a blockage that disturbs the ionic current through the pore, resulting in a measurable signal. These signals contain information about the structure, dynamics and others of the target molecules [50,51]. As materials, nanopores can be divided into biological nanopores [52,53] and solid-state nanopores [54,55], and they have contributed to probing different DNA higher-order structures.
2.2.4. SM-SR microscopy
Conventional fluorescence microscopy is limited by diffraction, which defines the achievable resolution. SM-SR technology has enabled fluorescence microscopy to achieve a spatial resolution of approximately 20 nm by an order of magnitude. In this technology, fluorophores behave as point sources, and their image corresponds to the point-spread function (PSF) of the microscope. Controlling the emitting concentration allows all fluorophores emitted at the same time to be avoided; then, each molecule can be analyzed in the microscope image. The final super-resolution image can be reconstructed by locating the position of each single molecule through PSF (Fig. 3(c)) [56,57]. SM-SR microscopy has become one of the most efficient imaging methods at the nanoscale and is widely used in the field of DNA nanotechnology [58,59].
As the first far-field microscopic imaging technology to break the optical diffraction limit, stimulated emission depletion (STED) microscopy is a major category of SM-SR techniques. The basic principle of STED is to irradiate the sample with two laser beams at the same time. One laser beam is used to excite the fluorescent molecules, bringing the fluorescent molecules in the center to an excited state. After excitation, the fluorescent molecules undergo vibrational relaxation to the lowest state. Meanwhile, another beam quenches the excited fluorescent molecules outside the central region, causing the excited fluorescent molecules to return to the ground state through STED without the spontaneous emission of fluorescence. In this way, a final high-resolution image is obtained beyond the diffraction limit [60].
《Fig. 3》

Fig. 3. Schematic overview of single-molecule detection used to detect DNA structures. (a) smFRET; (b) Nanopore; (c) SM-SR microscopy. TIRF: total internal reflection fluorescence; t1, t2: different time point.
《2.3. The combination of single-molecule detection and manipulation methods》
2.3. The combination of single-molecule detection and manipulation methods
For developing the temporal and spatial resolution, combining single-molecule detection with a manipulation method introduces complementary advantages and provides more details in detecting DNA conformational changes. These methods show promising potential for the kinetic or thermodynamic analysis of dynamic DNA. Early efforts to combine optical trapping with singlemolecule fluorescence were first demonstrated by Funatsu et al. [61] to directly detect the interactions between a kinesin molecule and a microtubule. After that, it became more common to apply a combination of optical/magnetic tweezers and single-molecule fluorescence or FRET to DNA measurements [62,63], such as nucleosome unwrapping [64] and determining the conformational diversity of G4 [65]. The combination of optical tweezers with fluorescence microscopy and confocal microfluidics has been widely used. This technique subjects individual molecules to different reaction mixtures and provides information such as identity, conformational dynamics, and spatial dynamics [66,67]. In addition, Lee et al. [68] developed a method combining an optical trap with three-color FRET for measuring the structural transition or folding/ unfolding dynamics of more complex samples (e.g., Holliday junctions and double hairpins).
《3. Single-molecule techniques for characterizing different DNA structures》
3. Single-molecule techniques for characterizing different DNA structures
《3.1. DNA primary structure》
3.1. DNA primary structure
The primary structure of DNA is ssDNA, which is formed by arranging deoxynucleotides with phosphodiester bond linkages, without hydrogen (H)-bonding interactions. ssDNA is an important intermediate in biological processes, such as DNA replication, transcription, and repair [69–71]. In vivo, ssDNA undergoes dynamic conformational changes due to its flexibility; thus, measurements of these mechanical properties may provide clues to understanding the folding structures of DNA. Because of the high flexibility of ssDNA, the conformations of ssDNA can be described only statistically in a bulk assay. The mechanical properties of ssDNA have been widely explored by means of single-molecule methods [72] such as smFRET, fluorescence correlation spectroscopy, and force spectroscopy. Using these methods, the persistence length (an important parameter characterizing flexibility [73]) of ssDNA was found to range from 1.5 to 5.0 nm, influenced by the DNA sequence, contour length, and concentrations of cations in the buffer [74]. However, most of these measurements were conducted using long ssDNA (> 100 nucleotides (nt)), as it is more difficult to detect shorter ssDNA. Recently, a study investigated the flexibility of DNA with less than 14 nt, using smFRET. The results showed that ssDNA is flexible even when it is shorter than the persistence length [69].
Two factors affect the elasticity of ssDNA: base-stacking interactions and the sugar pucker conformational transition. A previous study measured and analyzed the elasticity of two ssDNA homopolydeoxynucleotides—namely, poly(dA) and poly(dT)—by means of AFM [72]. Compared with poly(dA), which was found to have an expected entropic elasticity behavior, poly(dT) showed two overstretching transitions in the FEC at about 23 and 113 pN, caused by base-stacking interactions and sugar pucker conformational transitions (from the C3' -endo pucker to the C2' -endo pucker), respectively. The sugar pucker conformational transition was also found in long ssDNA with a random sequence [75], and the energy difference between the two sugar pucker conformations was found to be 2–1674 J·mol–1 .
Temperature may also affect the extension of ssDNA. When ssDNA is stretched by a relatively low force (<10 pN), the extension of the ssDNA increases as the temperature increases, due to secondary structure formation. In contrast, when the stretching force is greater than 10 pN, the extension of ssDNA decreases with increasing temperature, indicating that temperature plays an important role in the elasticity of ssDNA [76,77].
As mentioned earlier, the interaction between a nanopore and a DNA strand produces changes in the ion current passing through the nanopore, making nanopores useful for DNA sequencing. As the most promising third-generation sequencing technology, nanopore technology offers several advantages such as high accuracy, low cost, and long read length.
《3.2. DNA secondary structure》
3.2. DNA secondary structure
3.2.1. Double-strand DNA
3.2.1.1. Elasticity, unzipping, and shearing force of dsDNA. Watson, Crick, and Franklin discovered the double-helix structure of DNA in 1953 [78,79]. Since then, the chemical and physical properties of double-helix DNA have attracted great attention, especially the elasticity of dsDNA. Previous studies have shown that the freely jointed chain (FJC) model and freely rotating chain (FRC) model are best suited to describe the elasticity of ssDNA [80–82]. The FRC model is essentially the same as the FJC model, except that the former fixes the bond angles between nearest neighbor monomers [83].

where kB is the Boltzmann constant, x is the average extension of the chain at a given stretching force F,  is the rotating unit length of the polymer chain, T is the absolute temperature, and L is the contour length.
is the rotating unit length of the polymer chain, T is the absolute temperature, and L is the contour length.
For dsDNA, the force-extension relationship becomes nonlinear under large forces. This behavior can be explained by the FJC model, while the wormlike chain (WLC) model is suitable for medium forces [84,85]. In the classical FJC model, the chain is represented as inelastic segments that are freely jointed [86]; in the absence of external forces, the directions of these segments are irrelevant [37]. In this model, the elastic response of the molecules is pure entropy [87]:

where  is the Kuhn length.
is the Kuhn length.
As an alternative model, the WLC (as the name suggests) is a polymer molecule that is viewed as a long, thin worm; the configuration of the polymer chain is a spatial curve that can be bent at every point along the whole chain. The WLC model was proposed in 1949 [88] and was applied to describe the elastic response of a single dsDNA molecule in 1995 [89]. The force F is related to the fractional extension (x/L) as follows [89]:

where A is the persistence length.
To extend the range of the formula’s applicability, Wang et al. [90] added a stretch modulus to modify the formula; the force F is now related to the fractional extension (x/L) by the following:

where K0 is the elastic modulus.
In 1992, Smith et al. [37] measured the elasticity of a single dsDNA molecule, in what was the first single-molecule study of nucleic acids. They simulated the movement of the DNA molecules attached to the beads under hydrodynamic and magnetic forces. To date, single-molecule manipulation techniques have been increasingly applied in nucleic acid research, and important progress has been achieved in this technology. In such techniques, one end of a dsDNA molecule is attached to a glass surface, while the other end is connected to a magnetic bead. DNA molecules are linked to different surfaces via specific reactions such as biotin–streptavidin or digoxin/anti-digoxin antibody. At stretching forces lower than 5 pN, DNA shows an elastic response dominated by entropic effects; at forces greater than 5 pN, enthalpic contributions play a key role. Previous studies have shown that torsionunconstrained B-form DNA (B-DNA) undergoes an overstretching transition when a shearing force of approximately 65 pN is applied [38,91], leading to an approximately 1.7-fold elongation of its original contour length. At 150 pN, DNA splits into single strands and completely recombines when relaxed [92].
Essevaz-Roulet et al. [93] and Bockelmann et al. [94] performed the mechanical separation of a single bacteriophage  DNA molecule and found that DNA mechanical unzipping occurred in the range of 10–15 pN. The change in force was found to be highly correlated with the DNA guanine/cytosine (GC) content, with the GCrich region having a higher unzipping force than the adenine/ thymine (AT)-rich region. This result is consistent with the findings of Rief et al. [92]. For poly(dG–dC) and poly(dA–dT) DNA strands, the unzipping forces are FG–C = 20 pN and FA–T = 9 pN. Based on the equilibrium measurement under different stretching forces, the unzipping/rezipping free energy and kinetics can be obtained [95].
DNA molecule and found that DNA mechanical unzipping occurred in the range of 10–15 pN. The change in force was found to be highly correlated with the DNA guanine/cytosine (GC) content, with the GCrich region having a higher unzipping force than the adenine/ thymine (AT)-rich region. This result is consistent with the findings of Rief et al. [92]. For poly(dG–dC) and poly(dA–dT) DNA strands, the unzipping forces are FG–C = 20 pN and FA–T = 9 pN. Based on the equilibrium measurement under different stretching forces, the unzipping/rezipping free energy and kinetics can be obtained [95].
3.2.1.2. Interaction between base pairs. In a DNA double helix, Hbonds connect the base pairs on the two strands, which helps with inter-strand stabilization, while base-stacking interactions occur between adjacent base pairs, providing both inter- and intrastrand stabilization (Fig. 4(a)) [96]. If the stacking free energy between two bases is too high or too low, the genome will be over-stable or unstable, which will affect the unwinding of the DNA during replication or transcription, leading to genetic instability [97].
《Fig. 4》

Fig. 4. Overview of single-molecule methods used to explore the interaction between base pairs. (a) Stacking interaction between base pairs. Reproduced from Ref. [99] with permission. (b) Schematic illustration of probing the base stacking forces using optical tweezers and DNA origami structures. Orange tether represents a single strand, red and blue represent terminal base pairs. Reproduced from Ref. [99] with permission. (c) Schematics of exploring the interaction between nucleotides by AFM. PEG: polyethylene glycol; H1: dsDNA helical structure. Reproduced from Ref. [100] with permission.
As early as 2004, the Sattin et al. [98] explored stacking interaction forces and obtained the force contribution of A–T or G–C base pairs. They found that, although the number of H-bonds of A–T and G–C were different, these forces were approximately equal. In addition, they reported that base stacking contributes more to dsDNA interactions than H-bonding. Kilchherr et al. [99] further explored the stacking forces (Fig. 4(b)) between DNA base pairs using optical tweezers. They prepared four base-pair stacking configurations created by the bases A and T and used optical tweezers to measure the force-extension data. After calculation, the free energy increments of each stack were found to be between –3.4 and –14.23 kJ·mol–1 —information that now guides the design of DNA-based devices.
Long-range (15–25 nm) interaction also occurs between base pairs. Using AFM, Luo et al. [100] calculated the long-range interaction between dissociated complementary base pairs (Fig. 4(c)) and found that that of A–T was (2.3 ± 0.2) pN and that of C–G was (3.5 ± 0.2) pN; these forces are attributed to the interactions of multiplex H-bonds of ordered water structures between nucleotides. This result may be important for understanding the DNA hybridization process.
3.2.2. The Holliday junction
The Holliday (four-way) junction is a four-stranded DNA intermediate in DNA recombination, which can undergo conformational transition and branch migration under specific conditions (Fig. 5(a)). In the absence of metal ions, Holliday junctions adopt a conformation of four helices pointing to square corners [101]. At Mg2+ concentrations exceeding about 50 μmol·L–1 , Holliday junctions exist as two different isoforms, both of which have a characteristic X-shaped architecture [102]. With an increase in Mg2+ concentration, the transition between the two conformations (ISO-I and ISO-II) becomes slower [103,104], and the rate of branch migration decreases rapidly [105,106]. Similarly, an external force can affect the conformational transitions of Holliday junctions. Using a technique that combined smFRET and optical tweezers, Hohng et al. [107] showed that the conformations of Holliday junctions are biased at 0.5 pN or lower (Fig. 5(b)). Nickels et al. [108] designed a DNA self-assembled force clamp (Fig. 5(c)) and showed that an external force can be applied to make Holliday junctions continuously switch between the two stacking conformations, ISO-I and ISO-II (Fig. 5(d)). These results illustrate the unique advantages of smFRET in exploring conformational changes between two Holliday junction states. Since single-molecule techniques can be used to observe the intermediates and isomerization of a Holliday junction in real time, these techniques can be applied to explore internal characteristics and mechanical stretching. Thus, they are of paramount significance for future work in analyzing complex folds in biological systems and designing and validating complex DNA nanocomponents.
《Fig. 5》

Fig. 5. Holliday junction dynamics probed with smFRET experiments. (a) Schematic of branch migration and conformational transition of Holliday junctions. Cy5 and Cy3 fluorophores are terminally attached to two arms, respectively. (b) Optical tweezers assay for probing the conformational transitions of Holliday junctions. Holliday junction is tethered to the surface through biotin. Reproduced from Ref. [107] with permission. (c) Schematic of DNA force clamp. Connect the molecular system of interest (red rectangle) with the two anchor points of DNA force clamp. Reproduced from Ref. [108] with permission. (d) Conformational transitions of the Holliday junction under force. Reproduced from Ref. [108] with permission. F: force.
3.2.3. The DNA G4, i-motif, and triplex
G4s are four-stranded DNA secondary structures composed of folded guanine-rich nucleic acid sequences [109]. A G-quartet is a structural unit of G4s, and two or more layers of quartets form quadruplexes by means of π–π stacking. Scientists first identified G4s in cancer cells in 1962, finding that their stabilization can effectively inhibit the proliferation of tumor cells [110]. G4 folding topologies are believed to be divided into three types—namely, hybrid-stranded structures, antiparallel-stranded structures, and parallel-stranded structures (Fig. 6(a)) [111]—which are determined by two factors: the glycosidic conformations and the relative strand orientations [112]. The mechanical stability of these structures can be explored via magnetic tweezers. Cheng et al. [113] found that all non-parallel-stranded G4s show an unfolding force peak <40 pN, while parallel-stranded G4s show an unfolding force peak in the range of 40–60 pN, indicating that parallelstranded G4s have high mechanical stability. When the human telomere G4 is induced by K+ , it exists in a mixed conformation [114]. The conformational change kinetics of G4 can usually be detected by means of smFRET [115–117]. Long and Stone [118] found that in situ refolding leads to a dynamic distribution of the telomere DNA G4 conformation. The length and sequence of the loop also affect the G4 conformation and dynamics. Recently, the conformational dynamics of the G4 structure were also observed in the presence of Na+ . Noer et al. [119] identified at least four FRET states, which indicates that telomere G4s polymorphism does not only occur in the presence of K+ ions. As a label-free singlemolecule method, nanopores have also been employed to monitor the folding/unfolding dynamics of G4 [55,120,121]; they exhibit potential for higher spatial and temporal resolution than other methods.
《Fig. 6》
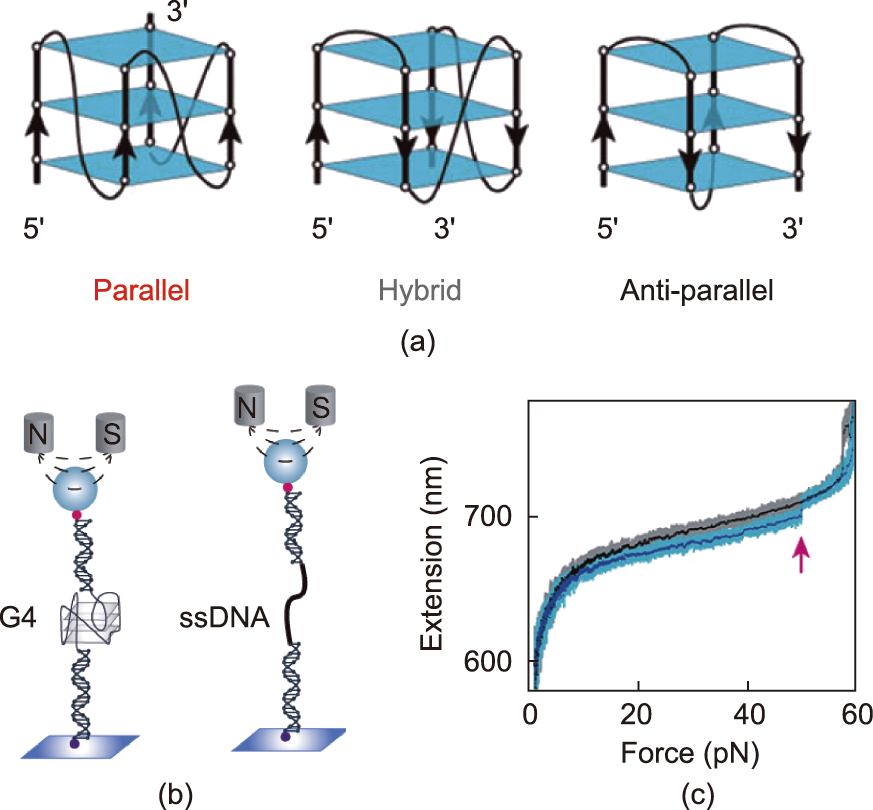
Fig. 6. Stretching of G4 using magnetic tweezers. (a) Three G4 folding topologies. Reproduced from Ref. [113] with permission. (b) Experimental set up of magnetic tweezers measurements. (c) The force-extension curves. The red arrow points to G4 unfolding. Reproduced from Ref. [123] with permission.
You et al. [122] has made efforts to explore the mechanical properties of the G4 with magnetic tweezers (Figs. 6(b) and (c)). They reported three folding states of telomeric G4, which have distinctly different mechanical stabilities and lifetimes. You et al. [123] studied the DNA G4 formed in the promoter region of oncogene c-myc and found that the unfolding rate of its main species is slow, which may be the reason for the gene-silencing function of c-myc G4 [122,124]. Another G-rich region—namely, the P1 promoter of the human B-cell lymphoma 2 (Bcl-2) gene—was also investigated. In this region, multiple G4 structures can be formed. Two major species, Bcl2-2345 and Bcl2-1245, were also studied due to their complex folding/unfolding kinetics [125]. Through an exploration of the folding/unfolding dynamics, the researchers found that the mechanical stability of Bcl2-2345 G4 was lower than that of Bcl2-1245 G4. This information may guide the design of G4-targeted small molecules that can regulate Bcl-2 gene expression in the future. Compared with canonical G4s, noncanonical G4s have been less explored. Zhang et al. [126] reported that G4-forming sequences with bulges can form a fully folded G4 (high mechanical stability) and partially folded intermediates (low mechanical stability), which depend on the lengths and positions of the bulges.
The KIT gene is located on human chromosome 4q12–13 and belongs to the proto-oncogene, which encodes the stem/mast cell growth factor receptor gene c-kit. Abnormalities in the c-kit gene may lead to abnormal cell increases and tumors [127]. There are three different G4 structures within the proximal promoter of KIT gene: kit*, kit1, and kit2 [128]. Previous studies have shown that the G4 formed at these sites will affect gene expression [129]. Understanding the conformational characteristics and mechanical properties of these G4 structures and deeply exploring their regulatory role in DNA structure is of great significance for taking them as potential drug targets. Buglione et al. [130] found that the formation of a G4 did not affect the FEC. When a fixed negative torsional stress was imposed (negative supercoiling = –40), wild type c-kit was extended under a low force (F = 0.3 pN, forming plectonemes), but mutated c-kit was not. Under a high force (F > 1 pN, forming denaturation bubbles), the curves of mutated c-kit and wild type c-kit almost coincide, close to the dsDNA extension curve under relaxation; in other words, the existence of a supercoil has a great influence on the mechanical properties of DNA in the c-kit region.
《Fig. 7》
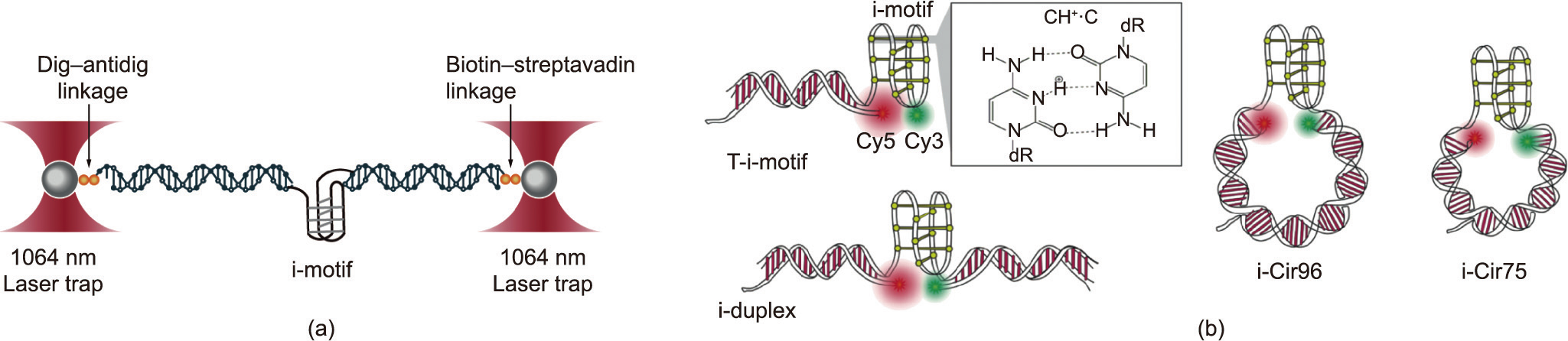
Fig. 7. The mechanical properties of i-motif probed using optical tweezers and smFRET. (a) Optical tweezers assay for stretching the i-motif. (b) Schematics of four different DNA origami structures. DNA constructs with the i-motif are connected at the terminal position of a DNA duplex (T-i-motif) or embedded in DNA duplexes(i-duplex) and DNA nanocircles (i-Cir96 and i-Cir75). Green and red indicate Cy3 and Cy5 fluorophores, respectively. Reproduced from Ref. [139] with permission.
Similar to the G4, the i-motif has been simulated since the 1990 s, but it has gradually attracted the attention of scientists in recent years. The i-motif is a four-chain structure formed by two parallel double strands by inserting hemiprotonated cytosine base pairs (C/C+ ) [131], it is widely found in genomic DNA [132,133] and stabilized in slightly acidic pH [134]. Several findings have demonstrated that i-motif sequences play a vital role in a variety of biological processes such as the regulation of gene expression and replication [135–137]. Dhakal et al. [138] was the first to explore the mechanical properties of the i-motif at the single-molecule level (Fig. 7(a)). These researchers determined that the i-motif had rupture forces of 22–26 pN. Using combined smFRET and self-assembled DNA nanostructures (Fig. 7(b)), Megalathan et al. [139] demonstrated that the human telomere sequence remains unstructured at pH 9.0 and adopts a fully folded i-motif sequence structure at pH 5.5. However, at a weakly acidic pH (pH 6.5), the sequence undergoes switching kinetics between the i-motif, partially folded state, and fully unfolded state. Topological constraint has also been shown to affect the folding and conformational dynamics of the i-motif. On this basis, Megalathan et al. [140] investigated the effect of molecular crowding on the stability of i-motifs embedded in nanocircles. To simulate the real topology of i-motif structures in chromatin in vivo, the researchers constructed ligated DNA nanocircles i-Cir96L (where ‘‘L” means ligated) (Fig. 7(b)). Polyethylene glycol (PEG) was used to mimic molecular crowding. Compared with that in unligated i-Cir96 nanocircles, the i-motif in ligated constructs formed only at pH 5.5, while the former showed significant folding at pH 6.5 and a large fraction of the partially folded state at pH 7.0 and 7.4 [139,141], indicating that the ligated nanocircles became hard so that DNA bending destabilized the i-motif. In the presence of PEG, even at pH 7.4, the i-motif showed a high FRET state, indicating that PEG can stabilize the i-motif.
The α-hemolysin protein nanopore was applied by the Ding et al. [142] to study the folds of the i-motif structure for the first time. By encapsulating the i-motif structure into a nanocavity, they reported a method for analyzing the lifetimes of the i-motif. The results showed that the folding lifetime of the i-motif decreased with increasing pH. Xi et al. [143] further found that changing the loop sequence and length did not affect the formation of the i-motif structure, although a longer loop length decreased the thermal stability of the i-motif. Without force, Jonchhe et al. [144] measured the half-life of the i-motif to be about 3 s at neutral pH using microfluidic channels. These methods are novel platforms for researching single-molecule kinetics.
A DNA triplex forms when a pyrimidine or purine base forms a Hoogsteen pair with the purines of Watson–Crick base pairs and occupies the main groove of a DNA double helix, and triplexforming oligonucleotides form intermolecular triplex strands with the target sequence on dsDNA (Fig. 8(a)) [18]. A triple helix may have many different compositions and geometries [145]. Ling et al. [146] reported a DNA triplex with 30 triads and determined that the force for rupturing the double strand and the third strand was approximately 42.6 pN. Lee et al. [147] investigated the formation of a DNA triplex by means of smFRET (Fig. 8(b)). When the pyrimidinemotif triplex was in a weak basic condition (pH 8.5), the singlestrand tail was not folded. With an increase in acidity, the low FRET efficiency peak disappeared and a new peak appeared at high FRET efficienc, indicating the formation of a parallel triplex. The presence of Mg2+ can also help the formation of a triplex, whether a purine– motif triplex or a pyrimidine–motif triplex. Further insight into the dynamics of forming a triplex came from Li et al. [148], who used a single-molecule rescue-rope-strategy and found that the formation of a DNA triplex also depended on the DNA sequence. The AA mutant (changing the motif of TTAGGG to AAAGGG) contributes to the probability of the formation of a DNA triplex structure.
《Fig. 8》
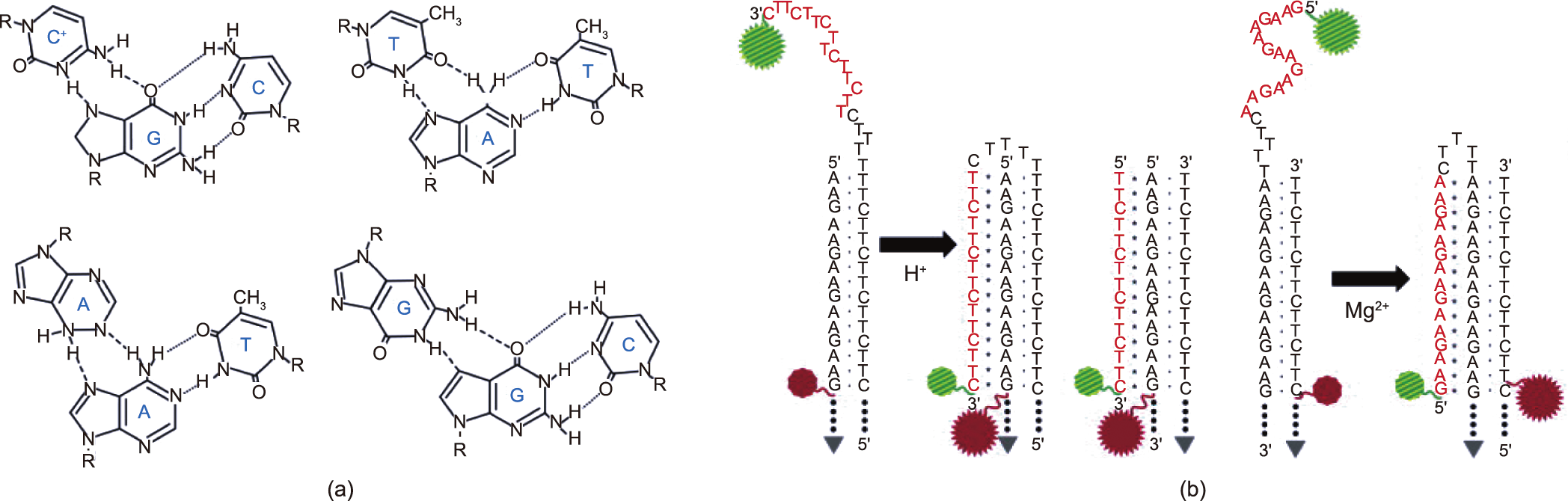
Fig. 8. DNA triplex. (a) Four base arrangements in DNA triplexes. Reproduced from Ref. [147] with permission. (b) Molecular constructs of pyrimidine–motif triplex (two on the left) and purine–motif triplex (right). Once it is folded, two fluorophores approach, resulting in a high FRET efficiency that can be observed. Reproduced from Ref. [147] with permission.
《3.3. DNA tertiary structure》
3.3. DNA tertiary structure
3.3.1. DNA topology
The DNA double helix causes torsional stress in the process of replication and transcription, which creates supercoils and causes topological changes. The DNA topology is described by the linking number (Lk), where Lk = twist (Tw) + wrist (Wr) [149], and where Tw refers to the number of helix turns in DNA and Wr is the number of times the double helix crosses itself (i.e., the number of supercoils). This intertwined structure is called a plectoneme [150]. In general, both negative and positive supercoils are generated in vivo. As the replisome advances, positive supercoils accumulate in front of the replication fork, which causes the replisome to rotate to relax the supercoils, allowing the two daughter DNA strands to become entangled [151]. When DNA transcription occurs, positive supercoiled DNA is produced before the transcription bubble and negative supercoiled DNA is produced after the transcription bubble [152]. Such a locally entangled DNA strand is necessary for transcriptional activation [153].
Single-molecule magnetic tweezers have become an effective tool for studying supercoiled DNA, because they can easily introduce superhelical turns by applying rotation to the beads using magnets. When negative torsional forces are applied to DNA, the torsional stress is usually stored in plectonemes rather than twists at low forces (< 0.5 pN). As the force increases, the DNA will start to twist, and the change causes DNA denaturation to form lefthanded structures [154,155]. When the force increases to 2 pN, the negative torsional force is completely absorbed by the denatured structure, and the DNA length does not change [156]. Strick et al. [154] tested six DNA sequences with different GC contents (Fig. 9(a)) and obtained an extension supercoiling density curve (Fig. 9(b)). Below 0.5 pN, the curve was similar to each other; above 0.5 pN, because of partial melting of the DNA, the DNA length increased, and the increased length of different sequences was different. DNA molecules with a higher GC content showed greater DNA extension. Kim et al. [157] explored the B-form DNA to Zform DNA (B–Z) transition kinetics of thymine guanine (TG) repeats; their smFRET results showed that TG repeats are sensitive to torsion and tension. The rates and rate constants of the B–Z transition were also calculated. In a previous work, Lee et al. [63] calculated the forward rate (KBZ = 0.051 s–1 ), reverse rate (KZB = 0.070 s–1 ), and equilibrium constant (Keq = KBZ/KZB = 0.73) of the GC repeat B–Z transition at 37 °C. In contrast, the transition rate of TG repeats is much higher, which shows that the free energy barrier between the two states is lower [156].
《Fig. 9》

Fig. 9. Applying torsion to explore DNA B–Z transition by single-molecule magnetic tweezers. (a) The schematics of single-molecule measurement on DNA supercoiling by magnetic tweezers. The supercoiling density-extension curve shows different DNA structures (denatured, underwound, or plectonemes). At a characteristic force (Fchar), negative supercoiling induces local melting of the dsDNA. σ: supercoiling density. Reproduced from Ref. [156] with permission. (b) The curves show the DNA extension as a function of supercoiling density under different forces. When the force is increased to 1.2 pN, all sequences with different GC contents show an asymmetric curve (asym.), with negative supercoiling being absorbed into DNA melting and positive supercoiling being absorbed into plectonemes. The final GC content of the 6 different DNA constructs were 77%, 58%, 42% (asymmetric distribution of GC along the molecule), 42% (symmetric distribution of GC along the molecule), 39% and 38%. Z: the DNA extension; LC: contour length. Reproduced from Ref. [156] with permission.
It is difficult to apply torsion to DNA with conventional optical tweezers. To overcome this challenge, Forth et al. [150], Sheinin et al. [158], and Deufel et al. [159] captured a nanofabricated quartz cylinder with optical tweezers, tethered the detected biomolecules between the bottom of the cylinder and the glass slides, manipulated the cylinder to stretch, twisted the biomolecules, and obtained the torsional modulus of DNA (Fig. 10(a)). Such an advanced device is called an angular optical trap (AOT). The researchers twisted a single-substrate DNA and braided a braided substrate DNA (i.e., braided two strands of DNA together) [160]. The results showed that, for naked DNA, the torsional modulus of ssDNA is three times higher than the torsional modulus of braided DNA, while for chromatin, the torsional modulus of braided chromatin is five times higher than the torsional modulus of single chromatin. In other words, when replication occurs on naked DNA, the supercoils produced by the replication will be mainly distributed behind the replisome (where the substrate is softer) (Fig. 10(b)), while the opposite occurs for the chromosome.
《Fig. 10》
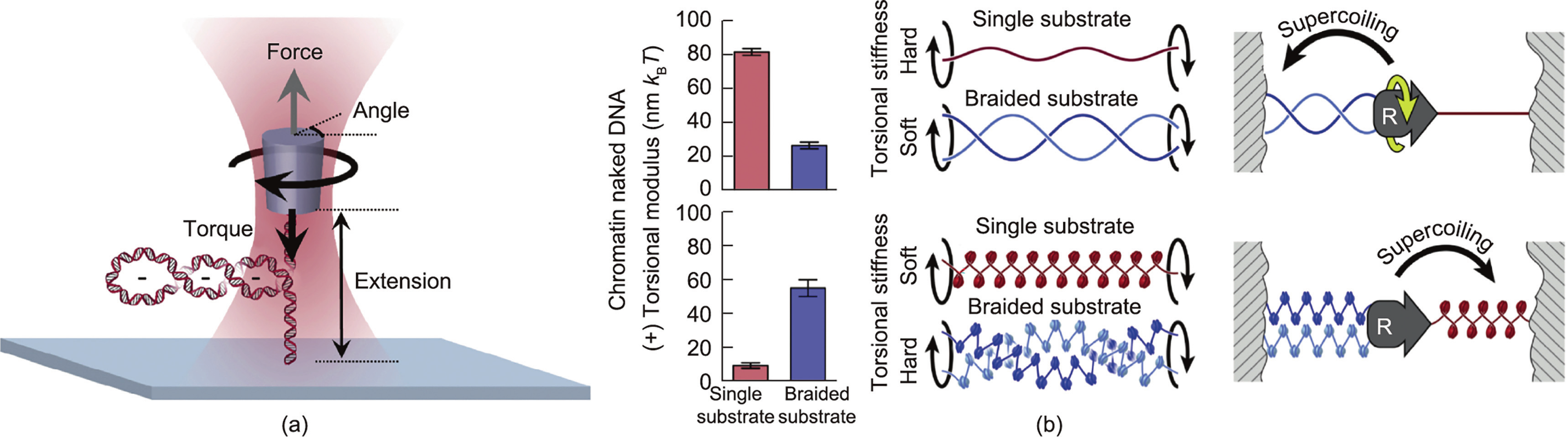
Fig. 10. Exploring DNA topology by angular optical trap (AOT). (a) Schematic diagram of AOT. Reproduced from Ref. [160] with permission. (b) The histogram of torsional modulus (left) of single (red) and braided substrate (blue) reveals the difference between DNA and chromatin substrates in the process of replication (right). Replication on naked DNA distributes supercoiling mainly to the behind of the replisome, while replication on chromatin is mainly located in front of the replisome. The kBT is 4.14 × 10–21 J, where kB is Boltzmann constant and T is the temperature. Reproduced from Ref. [160] with permission.
3.3.2. Nucleosome and chromatin
In eukaryotes, the genome is organized as chromatin, which is composed of DNA and histones. The basic unit of chromatin is the nucleosome, where 147 base pairs of DNA are wrapped around histone octamers (H2A, H2B, H3, and H4) for approximately 1.7 turns [161,162]. Chromatin is involved in all DNA metabolic processes, including transcription, replication, repair, and recombination. The potential of AFM for exploring the behavior and properties of individual chromatin strands has been demonstrated [163–165]; the formation of a higher-order chromatin structure can also be shown by AFM imaging [163]. Recently, Kilic et al. [166] revealed chromatin fiber interconversion kinetics using smFRET and found that heterochromatin protein 1αmaintains chromatin in a compact and dynamic state.
Single-molecule methods permit the observation of individual nucleosomes under stressed and torsional conditions, which is important for simulating physiological processes in vivo. Previous single-molecule studies have shown that nucleosomes will undergo a transition from partially unwrapped to fully unwrapped at forces of 15–25 pN [167–169]. When a force of approximately 10 pN was applied to the DNA template, the chromatin assembly process slowed down significantly [170]. Magnetic tweezers have been exploited to monitor nucleosome assembly on topologically constrained DNA molecules (Fig. 11(a)). Gupta et al. [171] found that positive supercoiling in the range of 0.025–0.051 will preclude nucleosome formation. Monoubiquitination at lysine 119 of histone H2A (ubH2A) can influence the stability of nucleosomes. Using magnetic tweezers, Xiao et al. [172] compared the unwrapped force of DNA with those of the H2A nucleosome and ubH2A nucleosome (Fig. 11(b)). They found that the ubH2A nucleosome underwent two-step unfolding at much higher forces. ubH2a can increase nucleosome stability by preventing DNA peeling from the histone octamer. Facilitates chromatin transcription (FACT) is a histone H2A–H2B dimer chaperone [173] that can facilitate the progression of polymerases on chromatin [173] and improve the fidelity of transcription [174]. FACT has also been shown to mediate nucleosome assembly and disassembly at the single-molecule level [175]. Recently, Wang et al. [176] directly studied the effect of ubH2A on FACT functions. By manipulating nucleosomes, ubH2A was discovered to regulate the dual functions of FACT in different ways. The function of FACT in nucleosome assembly is not affected by H2A ubiquitination; however, ubH2A greatly restricts the binding of FACT to nucleosomes, thus inhibiting its nucleosome disassembly activity.
《Fig. 11》

Fig. 11. Assembly and disassembly of nucleosomes probed using magnetic tweezers. (a) Assembly of the nucleosome using magnetic tweezers. A single dsDNA molecule is tethered between a magnetic bead and substrate surface, and then a mixture of core histone and histone chaperone Nap1 is added. The formation of nucleosomes is determined by monitoring the shortening in DNA extension (4Z) of the magnetic beads. Reproduced from Ref. [171] with permission. (b) The force-extension curves of H2Anucleosome and ubH2A-nucleosome. Reproduced from Ref. [172] with permission.
《4. The combination of single-molecule methods and DNA nanotechnology for detecting DNA structures》
4. The combination of single-molecule methods and DNA nanotechnology for detecting DNA structures
DNA nanoassemblies are widely used in nanomaterials, biomolecular sensing, imaging, and drug delivery [177–180]. Due to their high resolution, single-molecule techniques can be used to characterize the mechanical properties of individual nano-objects. Thus, the combination of single-molecule methods with DNA nanotechnology fills a research gap. There are numerous explicit reviews of DNA nanotechnology [181–183]. Here, we focus on two topics: ① using DNA nanostructures as static platforms to manipulate and observe single molecules; and ② studies of the structural dynamics of DNA nanostructures.
《4.1. DNA origami nanocages》
4.1. DNA origami nanocages
In 1991, Chen et al. [184] were the first to report the synthesis of closed polyhedral objects using DNA, after which DNA nanocages with different functionalities, shapes, and sizes were gradually constructed [185–187]. These nanocages exhibit extreme stability, making them useful for single-molecule studies. Nanoconfinement can increase the stability of the secondary structure of DNA or proteins due to entropic effects, such as G4s and imotifs. Shrestha et al. [188] used optical tweezers to explore the mechanics of G4s in DNA origami nanocages (Fig. 12(a)). They first used a medium nanocage construct (9 nm × 9 nm in cross-section) and found that the unfolding force of the G4s was significantly higher than the same sequence without a nanocage. Different nanocage sizes also affected the folding of G4s, with smaller sizes resulting in greater unfolding forces. Jonchhe et al. [189] found that the DNA hairpin acts in the opposite way (Fig. 12(b)). Previous studies have shown that the increase in the unfolding barrier of G4s in nanoconfinement is due to the hydration of water molecules in the transition process [190]. The abnormal phenomenon of hairpin DNA inside a nanocage is attributed to reduced water activity. In addition, the researchers suggested that the interaction of cations with the negatively charged origami surface was also responsible for compromising the stability of the dsDNA.
《Fig. 12》

Fig. 12. Design of DNA nanocages. (a) Top: schematic of the nanocage containing a G4-forming sequence. Bottom: A G4 sequence is ligated with two dsDNA handles, the two ends are labeled with digoxigenin and biotin. Reproduced from Ref. [188] with permission. (b) Schematic of the nanocage containing a hairpin. Reproduced from Ref. [189] with permission.
《4.2. DNA origami frame》
4.2. DNA origami frame
Compared with magnetic tweezers and optical tweezers, AFM can directly image biomolecules. AFM makes it possible to directly observe the dynamic movement of enzyme–dsDNA interactions, although it is laborious to control the orientation of DNA strands [191]. To solve this challenge, Rajendran et al. [191] and Sannohe et al. [192] created an observation platform based on DNA origami, termed the ‘‘DNA frame,” which could carry substrate dsDNAs (Fig. 13(a)). The researchers combined DNA origami with highspeed AFM to visualize changes in DNA conformation [193]. In the presence of K+ , the two dsDNA bridges in the DNA frame distinctly showed an X-type structure, indicating the formation of G4s (Fig. 13(b)). In contrast, G4 broke without K+ . Intermediate states were also observed in the DNA frame, such as the Ghairpin and G-triplex (Fig. 13(b)). To summarize, the DNA origami frame can be used to observe the formation and destruction of the G4, which is the first direct visualization of the solution-state structure of intermediates in the G4 folding pathway. Later, Feng et al. [194] reported that nucleosomes were rejected when they were in close proximity, whereas the two nucleosomes at the distal end can remain in stable contact for long periods of time. The DNA frame has also been employed to visualize the B–Z conformational transition of dsDNA [195] and DNA structural changes [196]. Thus, an assembled DNA frame provides an effective platform to explore DNA and chromatin structures in vitro.
《Fig. 13》

Fig. 13. Direct observation of DNA conformational changes in DNA origami frame. (a) Two different DNA origami frames. Reproduced from Ref. [197] with permission. (b) AFM images of the dynamic formation of the G4s (top) and formation of G-hairpin and G-triplex intermediates (bottom). Reproduced from Ref. [197] with permission.
《4.3. Structural dynamics of the DNA nanostructure 》
4.3. Structural dynamics of the DNA nanostructure
Single-molecule force spectroscopy permits deep exploration of the assembly of DNA nanostructures. Optical tweezers experiments show that the effective density of the Holliday junction determines the mechanical stability of a DNA origami structure. Under the action of an external force, the mechanical isomerization of two conformations of DNA nanotubes can be observed [198]. Bae et al. [199] reported a strategy based on magnetic tweezers to control the mechanical folding of DNA origami. The scaffold DNA was mechanically stretched to remove the secondary structure, and short strands were then introduced. Subsequently, the force was quenched, and displacement between the staple strands caused the folding of the DNA nanostructure. The entire folding process occurred within ten minutes. These investigations demonstrate the superiority of single-molecule force spectroscopy in probing the mechanical properties of DNA nanostructures.
Single-molecule FRET studies can monitor the reversible reconfiguration of DNA nanostructures. Saccà et al. [200] designed a reconfigurable DNA nanochamber, which changed the inner cavity size within DNA origami structures. Many recent studies have used DNA nanostructures as drug-delivery carriers. The DNA tetrahedron is a stable nanostructure for drug delivery. Goodman et al. [201] reported the conversion of a closed tetrahedron (high FE) to an open tetrahedron (low FE)—a process that can initiate the drug-release mechanism. A DNA box with a hollow cavity is another typical example. The lid can open depending on the specific oligonucleotide, and this process can be measured by FRET spectroscopy [202]. With a combination of Brownian dynamics simulations and smFRET microscopy, Jepsen et al. [203] found that Mg2+ concentration affects the structural rigidity of the DNA box. These studies provide insight into improving the stability of DNA nanostructures and optimizing their functions.
《5. Conclusions and prospects》
5. Conclusions and prospects
In conclusion, the inter- and intra-strand interactions of DNA lead to the formation of higher-order structures of DNA. Understanding the chemical and biophysical properties of different DNA higher-order structures is essential in uncovering their biological functions and for the design and construction of DNA nanostructures. With the aim of deciphering the properties of higher-order structures, many static structure-detection methods such as TEM, cryo-EM, and X-ray diffraction have been used to determine the structures of DNA. By analyzing the data from the structure, parameters such as persistence length can be calculated and obtained. In addition, high-speed AFM imaging and NMR can directly measure the dynamics of DNA conformational transitions. However, high-speed AFM imaging requires the sample to be attached to the surface and thus cannot entirely imitate the liquid environment. Although NMR can be used to detect the dynamic conformational transition, its temporal resolution is a few seconds [204]; thus, NMR cannot record fast dynamic structural transitions in real time. In contrast, single-molecule detection and manipulation methods can assist in detecting the conformational changes of DNA with faster transition kinetics, such as the transition kinetics from the unfolded state to the folded state of hairpin DNA [95]. The high temporal and spatial resolution of single-molecule methods provides a distinguishable signal to detect conformational changes. Another advantage of single-molecule manipulation methods is that these methods can directly stretch and manipulate a single DNA molecule, which enables detection of the elasticity of DNA. The applied forces also decrease the transition energy barrier from different states and accelerate the transition kinetics. Hence, single-molecule methods provide a powerful detection tool for detecting higher-order DNA structures.
With the rapid development of single-molecule techniques, these methods have also been adopted in the field of dynamic DNA nanotechnology. Combined with single-molecule detection and strand displacement reactions [205], applications including the detection of DNA glycosylases [206], characterization of DNA origami microarrays [207], and DNA navigators [208] have emerged. We anticipate that more effective single-molecule characterization methods will arise. Recently, Pei et al. [209] introduced a resettable DNA computing method based on singlemolecule magnetic tweezers and a toehold-mediated DNA strand displacement reaction. Previously, Koirala et al. [210] and Mandal et al. [211] also presented several DNA detection methods involving single-molecule optical tweezers. In this direction, we expect various DNA storage, DNA detection, and DNA computing methods to be developed in the future.
Although extensive studies of DNA structure have been conducted, the potential and functions of endogenous and exogenous modified nucleic acids still require further investigation. In vivo, epigenetic modifications of DNA or DNA lesions may induce gene silence or mutations and may subsequently influence the fate of cells [212,213]. Studies have shown that the physical properties of modified DNA differ from those of DNA. Using magnetic tweezers detection, Yang et al. [214] suggested that the methylation of cytosine may enhance dsDNA condensation. McCauley et al. [215] provided biophysical evidence of the oxidation of a guanine base (oxoG)-induced mutation effects by means of a hairpin unzipping assay using optical tweezers. Artificial nucleic acids (also called xeno nucleic acids (XNA)) such as α-L-threofuranosyl nucleic acid (TNA), locked nucleic acid (LNA), and 2' -deoxy-2' -fluoro-β-D-arabinonucleic acid (FANA) have also emerged in preclinical studies [216–218]. XNA can resist enzyme digestion in vivo; thus, the high biological stability of XNAs provides a fresh perspective on nucleic acid therapy, including RNA-mediated interference (RNAi), antisense oligonucleotides (ASO), and aptamers [219]. DNA conformational structures can be changed by altering the nucleotide into XNAs, and the stability of the structures can be affected by the special interactions between XNAs. Recent studies demonstrated that 2' -deoxy-2' -fluoro-arabinonucleosides (e.g., 2' F-araG) substitution stabilizes the propeller parallel G4, while 2' F-araC modification stabilizes the i-motif [220,221]. Single-molecule methods can provide more information for probing the mechanical properties and conformational changes of different structures of oligonucleotides containing XNAs, which is of great significance for understanding how XNAs work in vivo.
However, obtaining an XNA strand is more difficult than obtaining DNA. There are two efficient methods for the synthesis of XNA strands: solid-phase synthetic and enzymatic approaches [222]. For solid-phase synthesis, the XNA amidites are expensive and not readily available, and the efficiency of the synthesis may be much lower. In contrast, enzymatic synthesis can obtain much longer XNA strands. However, it is necessary to evolve a natural enzyme to specifically recognize the XNA (e.g., mutated Tgo polymerase Tgo-D4K for FANA extension) [223]. The evolved XNA polymerases should have a high synthetic rate and fidelity; thus, their evolution requires plenty of work screening the proper polymerase mutant. Therefore, conducting single-molecule experiments for detecting the structures of XNAs depends on the availability of XNA strands.
Several challenges remain to be resolved in detecting DNA structures using single-molecule methods. So far, studies at the single-molecule level have been carried out in vitro, especially those involving measurements using single-molecule manipulation methods. We can simulate the in vivo environment as much as possible; for example, we usually use PEG or dimethyl sulfoxide (DMSO) to simulate a crowder in vivo [140,224]. Nevertheless, the gap between in vitro and in vivo research urgently needs to be filled. Recently, Syrchina et al. [225] manipulated the nucleolar in the nucleoplasm using optical tweezers. Keizer et al. [226] illustrated a genomic locus manipulation technique inside the nucleus using magnetic forces. These applications exhibit the potential of manipulating the DNA structure in vivo. Furthermore, not all single-molecule techniques can be applied with high throughput, which is a limitation in using single-molecule techniques to solve practical problems. For optical tweezers, a single beam can be time-shared via acousto-optical deflectors to create multiple optical traps, which is an effective way to improve throughput. Developing high-throughput optical traps remains an important challenge for single-molecule analysis.
Given the development of single-molecule technologies over the past few decades, we anticipate that more DNA higher-order structures will be discovered, while highly accurate and highthroughput single-molecule characterization methods will emerge.
《Acknowledgments》
Acknowledgments
This work was supported by financial support from the National Key Research and Development Program of China (2021YFF1200200), the National Natural Science Foundation of China (22161132008), the Natural Science Foundation of Shanghai, China (19520714100 and 19ZR1475800), the Starry Night Science Fund of Zhejiang University Shanghai Institute for Advanced Study (SNZJU-SIAS-006), and the Natural Science Foundation of Zhejiang Province (LQ21C050001).
We thank all the research subjects for their participation. The Riken group reported the summary data. We would also like to acknowledge the skillful work of all medical staff at Xuanwu Hospital, Capital Medical University, Beijing, China.
《Compliance with ethics guidelines》
Compliance with ethics guidelines
Yonglin Liu, Tianyuan Bian, Yan Liu, Zhimin Li, Yufeng Pei, and Jie Song declare that they have no conflicts of interest or financial conflicts to disclose.











 京公网安备 11010502051620号
京公网安备 11010502051620号




