

《1. Introduction》
1. Introduction
Secondary amyloid A (AA) amyloidosis is a systemic metabolic disease, which is a complication induced by serum accumulation of serum amyloid A 1 (SAA1) protein that is over-secreted by hepatocytes in reaction to acute and chronic inflammation [1,2]. This leads to the deposition of AA amyloid fibrils in key organs, resulting in multiple life-threatening human diseases, such as severe chronic infection, tumorigenesis, liver and kidney failure, and the serious complication of rheumatoid arthritis (RA) [3–7]. RA is a chronic and systematic autoimmune condition that leads to bone destruction, long-term pain, serious complications, and eventual progressive disability, which affects the quality of life of RA patients and creates a significant social burden on our society [8]. Genetic factors play a crucial role in the initiation of RA. Around 60% of RA cases in twin patients is related to genetic inheritance [9]. However, causative genes leading to the pathogenesis of RA, as well as those that contribute to the severe lifethreatening complications of RA, remain unidentified, making early diagnosis and effective treatment of RA difficult. When RA coexists with AA amyloidosis, lethal outcomes have been reported [1,10], with infection and renal failure being the most typical causes of death [7]. However, monitoring SAA1 serum levels is not yet a routine practice in RA patients, and the risk of SAA1 dysregulation in RA patients is still largely unknown.
Previously, using the whole-exome sequencing method, we identified multiple novel genes that were enriched with deleterious variants in 58 RA patients’ peripheral blood mononuclear cells [11]. One novel deleterious mutation was in the SAA1 gene, creating an amino acid change in the SAA1.2 protein isoform. Five different isoforms of the SAA1 have been reported [12] as a result of extensive genome-wide association studies. Little is known about the SAA1.1 and SAA1.4 isoforms; however, previous studies have reported that SAA1.3 and SAA1.5 were associated with RA in several clinical cohorts [2,13,14]. The SAA1.3 isoform was reported to be closely related to AA amyloidosis and disease onset, as well as poor prognosis in a Japanese RA patient cohort [7]. In contrast, SAA1.5 was reported to have a limited ability to induce inflammation owing to the low secreting ability of pro-inflammatory cytokines [15]. To our knowledge, the biological roles of these SAA1 isoforms, including our newly identified SAA1.2 mutation, have not yet been comprehensively investigated.
Here, we aim to clarify the role of the SAA1.2, SAA1.3, and SAA1.5 alleles as a pathogenesis factor or an inhibitory factor in the development and outcome of RA. We optimized an experimental adenovirus infection protocol to successfully transfer gene constructs to the bone cartilage genome of mice. We then examined the biological and functional roles of these three SAA1 isoforms as functions of time. The results indicated that the SAA1.2 protein isoform expression aggressively induced both the initiation and progression of arthritis, which is a pivotal process of RA, and thus might provide new insights into the role of the SAA1 isoform in the progress of RA.
《2. Materials and methods》
2. Materials and methods
《2.1. Adenovirus vectors and animal experimental protocol》
2.1. Adenovirus vectors and animal experimental protocol
Three recombinant adenoviruses harboring the full-length complementary DNA (cDNA) of three different human SAA isoforms, SAA1.2, SAA1.3, and SAA1.5, were cloned in front of the enhanced green fluorescent protein (EGFP) reporter gene. Adenoviruses were generated with constitutive expression under the control of the cytomegalovirus promoter (VectorBuilder, USA). Female C57BL/6 mice (10–14 weeks old) were used in our studies. After one week of adaptive feeding, all mice were randomly and evenly divided into four groups (ten mice each), including SAA1.2, SAA1.3, and SAA1.5 adenovirus injection groups and the EGFP control group. The rear right knee joints were injected laterally with 1 × 108 plaque forming unit (pfu) of adenovirus containing either EGFP and or one of the three SAA1 isoforms. The animals were sacrificed at weeks 1, 2, 4, and 6 after injection. The study protocol was approved by the institutional committee for animal welfare. All animals included in our studies were housed under a 12 h light/ dark cycle and provided free access to standard rodent food (5053-PicoLab Rodent Diet 20; LabDiet, USA) and pure water.
《2.2. Micro-computed tomography (micro-CT) analysis》
2.2. Micro-computed tomography (micro-CT) analysis
Mice were anesthetized and gently tied on a CT scanning groove with knee joints scanned by a micro-CT scanner (Bruker, Belgium). Scans were conducted using a voxel size of 9 μm. The X-ray tube current was 441 μA, and the voltage was 43 kV. The severity of bone erosion was ranked with the radiological score system according to the method described previously [16]. Moreover, the three dimensional (3D) microstructural properties of bone were calculated using CT-Analyzer, the software supplied by the manufacturer. We also developed a new evaluation method to assess bone destruction, called the micro-CT score. This score contained five disease-related indexes of the micro-CT analysis results for the calcaneus, including trabecular bone mineral density (BMD), tissue mineral density (TMD), trabecular number, total porosity, and the trabecular volume rate. We determined that all five indexes (X) changed synchronously with the development of arthritis but to different extents. Therefore, we modified and normalized the formula as follows: (X – min)/(max – min) or 1 – (X – min)/(max – min). Then we averaged the five items to get the final micro-CT score that exhibited all five aspects of bones. Additionally, we set a bone destruction level based on the microCT score to evaluate the extent of damage more visually and directly.
《2.3. Histological grading》
2.3. Histological grading
Knee samples were fixed in 4% paraformaldehyde, demineralized in 10% ethylene diamine tetraacetic acid (EDTA) for four weeks, dehydrated, embedded in paraffin, sectioned sagittally, and stained with hematoxylin and eosin (HE). To assess synovial inflammation, histological scoring based on the reported critical criteria was modified [17]. Synovial hyperplasia, fibrosis, inflammation, articular cartilage destruction, and bone erosion damage were used as criteria for scoring (Table 1 [17]).
《Table 1》
Table 1 Qualitative grading system used to assess the degree of synovitis in the mouse knee joint. Adapted from Ref. [17] with permission of American College of Rheumatology, ©2010.

《2.4. Immunohistochemical (IHC) analysis》
2.4. Immunohistochemical (IHC) analysis
Paraffin-embedded samples were cut into 5 μm sections. The distribution of SAA1 was studied with antibodies prepared against human and murine SAA1, which also detected human SAA1 (Abcam, UK). After washing, the slides were incubated with streptavidin-conjugated horseradish peroxidase for 10 min (BioSite Histo Plus (HRP) polymer anti-mouse kit; Nordic BioSite, Sweden). The color was developed with diaminobenzidine, and the sections were counterstained with hematoxylin.
《2.5. Flow cytometric analysis》
2.5. Flow cytometric analysis
Examination of the change in T cell percentages was investigated by a CD4 and CD8 staining assay according to the manufacturer’s protocol [18]. The indicated number of mouse spleen and peripheral blood mononuclear cells were resuspended in 100 μL of 1 × staining buffer, and anti-CD4 and anti-CD8 fluorescencetagged antibodies were added to the cell suspensions. At least 100 000 events were collected for quantitative analysis by flow cytometry (BD FACSCalibur, USA).
《2.6. Real-time polymerase chain reaction (RT-PCR)》
2.6. Real-time polymerase chain reaction (RT-PCR)
Knee samples were dissected 5 mm from the femoral head and 5 mm from the tibial head and prepared free of surrounding muscles. The samples were pulverized under liquid nitrogen. RNA was extracted using TRIzol reagent (Invitrogen, USA). The concentration of RNA was examined by a NanoDrop 2000 system (ThermoFisher Scientific, USA). cDNA synthesis was performed with cDNA synthesis kits (Roche, Germany). The primers (GenePharma, China) used for cDNA amplification are listed in Table S1 in Appendix A. CT values were obtained using a ViiATM7 high-productivity real-time quantitative PCR system (Life Technologies, USA).
《2.7. Enzyme-linked immunosorbent assay (ELISA)》
2.7. Enzyme-linked immunosorbent assay (ELISA)
The concentrations of the SAA and inflammatory factors interleukin (IL)-6, IL-22, matrix metalloproteinase (MMP)-3, MMP-9, IL-17, IL-23, and granulocyte-colony stimulating factor (G-CSF) were measured in mouse serum using commercially available ELISA kits (R&D, USA). According to the manufacturer’s instructions, the blood samples were allowed to clot for 2 h at room temperature (24–26 °C) before being centrifuged for 20 min at 3000 revolutions per minute (rpm). The serum was removed and assayed immediately or after storage at ≤–20 °C. Both samples and standards were analyzed in triplicate.
《2.8. Western blot analysis》
2.8. Western blot analysis
Liver, kidney, and lung tissues were lysed with ice-cold radio immunoprecipitation assay (RIPA) lysis buffer (ThermoFisher Scientific). The protein samples were electrophoretically separated and transferred to nitrocellulose membranes. The membranes were incubated with primary antibodies at 4 °C overnight and incubated with a secondary fluorescent antibody for 1 h at room temperature. Bands were visualized on an LI-COR Odyssey scanner (USA). Primary antibodies human SAA1 were purchased from Abcam and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) from Cell Signaling Technology (USA).
《2.9. Statistical》
2.9. Statistical
analysis All the data are presented as the mean ± standard deviation (SD) or standard error of the mean (SEM) (indicated in each figure) of three individual experiments. Differences were analyzed by oneway analysis of variance using Graph Pad Prism 5.
《3. Results》
3. Results
《3.1. Adenoviral expression of SAA1 isoforms induced bone destruction of knee joints in mice》
3.1. Adenoviral expression of SAA1 isoforms induced bone destruction of knee joints in mice
To identify the roles of the three SAA1 isoforms in RA, we created a timeline for animal experiments (Fig. S1(a) in Appendix A). Adenoviruses carrying SAA1 alleles and an EGFP control were injected with a single dose into the articular cavity of the right knee in mice; CT scores were monitored, and tissues were collected at four time points (weeks 1, 2, 4, and 6 after injection). We observed no differences in body weight among the groups during this six-week period, suggesting that adenovirus infection may not induce toxicity (Fig. S1(b) in Appendix A).
Then, we analyzed changes including trabecular BMD, TMD, trabecular number, total porosity, and the trabecular volume rate using axial micro-CT in both legs. In the right leg, bone destruction occurred from weeks 2 to 4 and gradually recovered by week 6; the CT scores of the SAA1.2, SAA1.3, and SAA1.5 groups decreased by 27.8%, 26.7%, and 10.9%, respectively, in week 2, and were restored to 8.7%, 15.8%, and 5.8%, respectively, in week 6, compared to those of the EGFP group. For BMD, SAA1.2 showed an 8.7% decrease in week 2, and SAA1.2 and SAA1.3 decreased by 12.3% and 11.3%, respectively, in week 4, and were restored to normal values in week 6. Other parameters also showed the most significant change in week 2, except for TMD (Fig. 1). To visualize bone destruction, we reconstructed a 3D model of the mouse tibia and observed remarkable attenuation of the trabecular bone in the SAA1 groups, especially in the SAA1.2 group in week 4 (Fig. S2 in Appendix A). The bone destruction recovered by week 6, which was consistent with a decrease in the SAA1 level, indicating that stress reaction recovery had occurred.
《Fig. 1》

Fig. 1. Qualitative analysis of bone changes after adenoviral infection by axial micro-CT. (a) Trend changes of CT scores over time course for three SAA1 isoforms compared with the EGFP control group (n = 8 in each group). (b)–(f) BMD, TMD, trabecular number (Tb.N), total porosity (TOT), and trabecular volume rate (BV/TV) values of right (the injected side) knee joints in all groups over the time course (n = 8 in each group). Values are presented as the mean ± SEM. * represents for the p-value for the comparison of each SAA1 allele to the EGFP control group. *: p < 0.05; **: p < 0.01.
《3.2. SAA1 isoforms induced synovial and systemic inflammation》
3.2. SAA1 isoforms induced synovial and systemic inflammation
Histological scoring [17] and qualitative scoring of the degree of synovitis were used to assess synovial inflammation (Table 1). The arthritis scores increased in week 2 in SAA1.2 and SAA1.3 groups. In week 4, only SAA1.2 remained significantly different, suggesting that SAA1.2 may dominate human RA progression. After week 6, the arthritis scores were decreased (Fig. 2(a)). Only one week after adenovirus infection, all SAA1 induction groups exhibited inflammatory responses in the articular cartilage, as illustrated by increases in mononuclear cell infiltration and synovial cell expansion (HE, Figs. S3–S5 in Appendix A). In week 4, superficial synovial erosion appeared in the SAA1.2 group, with no significant change in EGFP-treated animals. IHC analysis confirmed SAA1 protein expression from week 1 until week 4 in the synovium, growth plate, and even deep into the bone (Figs. 2(b)–(e) and S3–S5).
《Fig. 2》

Fig. 2. Overexpression of SAA1 isoforms induces synovial inflammation and adenoviral-SAA1 infects multiple tissues in the knee joints. (a) Qualitative grading results for synovitis in mouse knee joints according to synovial cell hyperplasia, fibrosis, inflammation, articular cartilage destruction, and bone erosion in all groups at the indicated time points (n = 5 in each group). (b)–(e) Analysis of HE and IHC images of sections of knee joints from the EGFP, SAA1.2, SAA1.3, and SAA1.5 groups after four weeks of infection. The black arrow indicates superficial erosion of articular cartilage in the SAA1.2 group, the red arrow indicates the positive expression of SAA1 in synovial cells, the yellow arrow indicates the expression of SAA1 in the growth plate, and the blue arrow represents expression in bone tissue. Values are presented as the mean ± SEM.
《3.3. SAA1 isoforms induced T cell-mediated immune response and infiltration into articular joints and release of pro-inflammatory cytokines》
3.3. SAA1 isoforms induced T cell-mediated immune response and infiltration into articular joints and release of pro-inflammatory cytokines
RA is characterized by T cell infiltration in the synovial membranes [19]. Flow cytometric analysis of mouse spleens in week 2 showed that SAA1.2 induced a 10.9% decrease in CD8+ T cells and an 11.3% increase in CD4+ T cells (Figs. 3(a) and S6(a) in Appendix A), resulting in an increase in the CD4+ /CD8+ ratio. No changes in CD8+ or CD4+ T cells were observed in the spleen in other groups throughout the six-week study period. On the contrary, compared to EGFP-treated mice, all SAA1 isoforms induced changes in CD4+ and CD8+ T cells in the blood in week 4 (Figs. 3(b) and S6(b) in Appendix A).
《Fig. 3》

Fig. 3. SAA1 isoforms induced T cell-mediated immune responses. (a) Flow cytometric analysis of the percentages of CD4+ and CD8+ T cells and the fold changes of the CD8+ / CD4+ ratio in the spleen and blood in the SAA1 groups compared with the EGFP group at four different time points (n = 8 in each group). (b) Flow cytometry analysis of the percentage of CD4+ and CD8+ T cells of blood and spleen in three SAA1 groups compared with the EGFP group at four different time points (n = 8 in each group). Values are presented as the mean ± SEM.
RT-PCR confirmed successful gene integration and transcription in the bone cartilage throughout the experimental period. SAA1 message RNA (mRNA) levels were elevated in week 1 and gradually decreased from weeks 2 to 6, but the levels remained significantly higher than those of EGFP-treated animals (Fig. 4(a)). An increase in secreted-SAA levels in serum was also observed (Fig. 4(b)).
《Fig. 4》

Fig. 4. SAA1 isoforms induced SAA1 expressed in right articular joints and SAA release in blood circulation. (a) RT-PCR analysis of the expression of SAA1 in right articular joints. (b) ELISA analysis of the secretion of SAA in serum. (n = 5–6 in each group). Values are presented as the mean ± SEM. # represents for the p-value for the value change of each SAA1 allele over time. #: p < 0.05; ##: p < 0.01; *** p < 0.001.
RT-PCR and ELISA results showed that IL-6 and IL-22 expression was remarkably stimulated in week 1 at right leg and blood circulation of mice transfected with SAA1 isoform (Figs. 4(a) and 5), consistent with a recent report that robust production of IL-22 induces SAA1 production and inflammation [20], further explaining why SAA1.2 caused the most severe bone damage (Figs. 1(c) and (d)). Previous reports demonstrated that these cytokines could polarize T cells into different subsets of T helper cells [21], which further produces different cytokines [22]. Our results also showed upregulation of IL-17, IL-23 (Figs. S7(a) and (b) in Appendix A), tumor necrosis factor (TNF)-α, IL-1β, and G-CSF (Figs. 6 and S7(c) in Appendix A). Increased TNF-α or IL-17 could enhance matrix MMP production in articular bone degradation, which ultimately destroys the joint structure [23–25]. In line with this finding, intra-articular secreted and blood circulated MMP-3 and MMP-9 were activated after SAA1 adenovirus injection (Fig. 7). At the same time, the MMP inhibitor tissue inhibitor of metalloproteinases 1 (TIMP-1) (Fig. S8 in Appendix A) exhibited trends opposite to those of the MMP levels.
《Fig. 5》
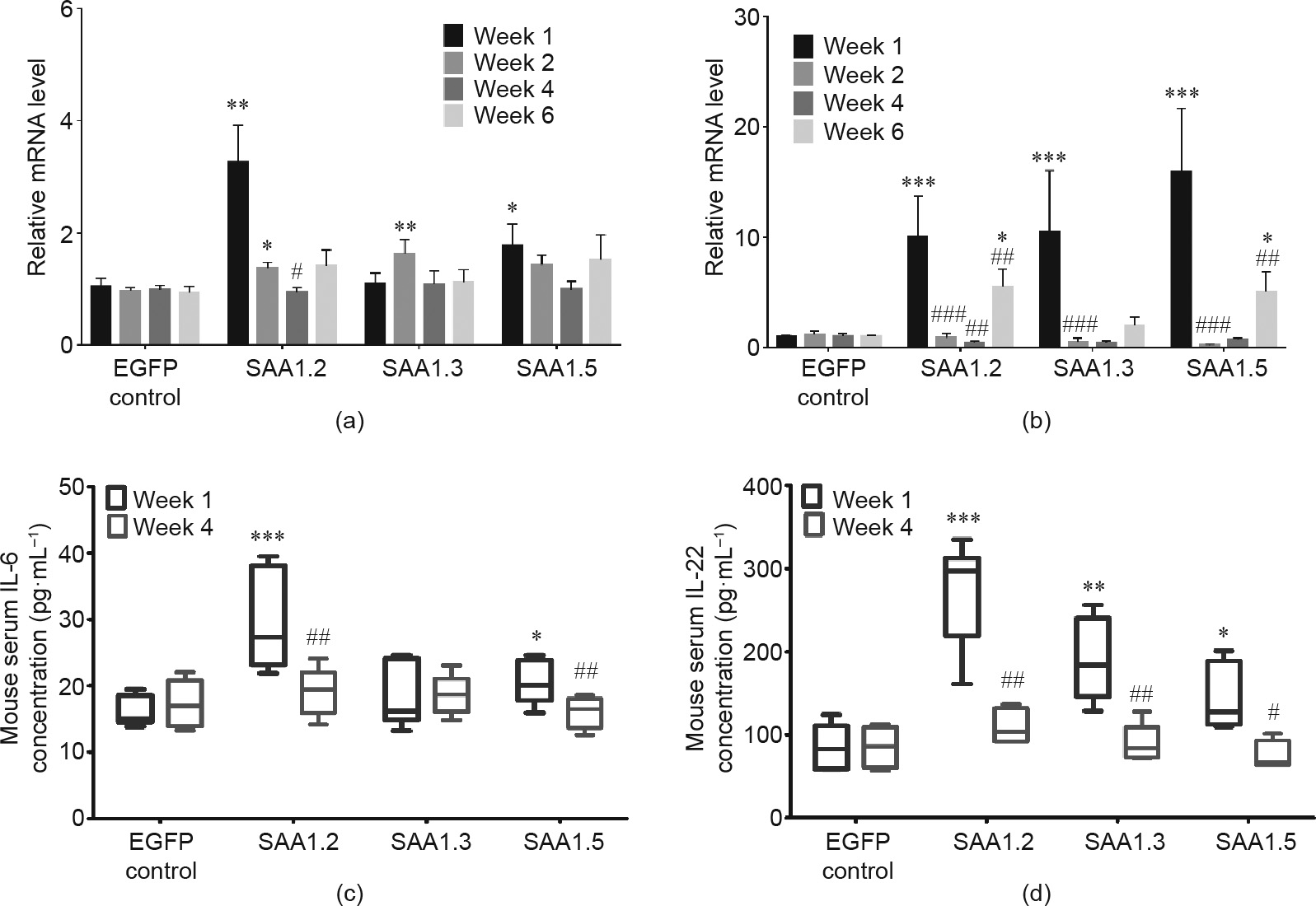
Fig. 5. SAA1 isoforms induced pro-inflammatory cytokines, IL-6 and IL-22, related mRNA expression in right articular joints and concentration in serum. (a) IL-6 related mRNA expression in articular joints; (b) IL-22 related mRNA expression in right articular joints; (c) IL-6 concentration in serum; (d) IL-22 concentration in serum. (n = 5–6 in each group). Values are presented as the mean ± SEM. ###: p < 0.001.
《Fig. 6》
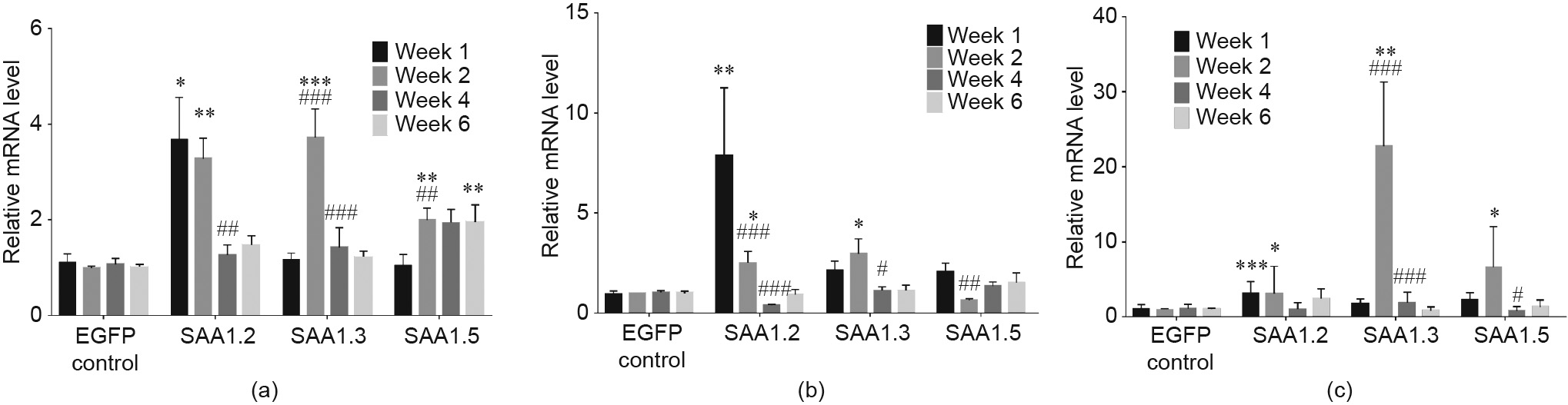
Fig. 6. SAA1 isoforms induced pro-inflammatory cytokines, TNF-α, IL-1β, and G-CSF, related mRNA expression in right articular joints. (a) TNF-α related mRNA expression in right articular joints; (b) IL-1β related mRNA expression in right articular joints; (c) G-CSF related mRNA expression in right articular joints (n = 5–6 in each group). Values are presented as the mean ± SEM.
《Fig. 7》

Fig. 7. SAA1 isoforms induced MMP-3 and MMP-9 related mRNA expression in right articular joints and concentration in serum. (a) MMP-3 related mRNA expression in right articular joints; (b) MMP-9 related mRNA expression in right articular joints; (c) MMP-3 concentration in serum; (d) MMP-9 concentration in serum (n = 5–6 in each group). Values are presented as the mean ± SEM.
《3.4. SAA1 isoforms induced left knee bone destruction and inflammation, and lead to SAA1 protein accumulation in key organs》
3.4. SAA1 isoforms induced left knee bone destruction and inflammation, and lead to SAA1 protein accumulation in key organs
Interestingly, all three SAA1 isoform protein expressions were also found in left knee joints on week 1, and the protein expression remained high until week 4 (Figs. S9 and S10 in Appendix A). There was no injection procedures were performed, and we did not observe significant bone erosion on the left knee joints biopsy (Figs. S9 and S10), but bone destruction was occurred. BMD showed significant decreases of 13.2% and 14.7% in the SAA1.2 and SAA1.3 groups, respectively, in week 4, and these values were restored to normal in week 6, suggesting that blood circulation of the SAA1 protein induces systematic inflammatory responses, as reported for human RA (Figs. S11(a), S12, and S13 in Appendix A). In addition, similar cytokine elevation was also observed in the left leg (Figs. S11(b) and S12 in Appendix A).
Surprisingly, we observed SAA1 protein accumulation in key organs, including liver, lung, and kidney (Figs. S11(c) and (d) in Appendix A). Further confirming that SAA1 circulates in the blood to the left leg and affecting other organs after expression at the right knee and induces the arthritis condition.
《4. Discussion》
4. Discussion
RA is a complex and difficult-to-treat autoimmune disease of unknown pathogenesis and characterized by the lack of a valid drug target [26]. Subclinical amyloidosis was commonly found in RA and other inflammatory rheumatic diseases that may lead to serious complications [27]. The prevalence rates of secondary amyloidosis in RA patients range from 7% to 26% [28], but thus far, the pathogenesis and consequence of amyloidosis with RA are largely unknown. Several SAA alleles were reported previously to be associated with RA; however, their pathological roles have not yet been clarified. SAA1.2 was first identified by us in RA patients using whole-exome sequencing [11,12]. The SAA1.3 allele has been reported to be a risk factor for the association of AA amyloidosis that adversely influences the outcome in RA patients. Fatal cases were reported in patients with SAA1.3 allele in a Japanese cohort, suggesting the potential importance of SAA1 single nucleotide polymorphisms [7]. SAA1.5 has been shown to exhibit slower serum SAA clearance compared to the other alleles, which indicates it has less amyloid genesis [15]. At the same time, SAA1.5 also has a limited ability to induce inflammatory cytokines than other alleles, which may weaken the inflammatory response [29]. We used the SAA1.3 allele as the positive control and SAA1.5 as the negative control to investigate the function of SAA1.2. In this study, the amino acid sequences of all SAA1 isoforms (Fig. S14 in Appendix A) were designed, we successfully clarified the role of these three SAA1 allele products to induce arthritis, which is one of the key symptoms of RA, and SAA1.2 was demonstrated to be the most aggressive.
Classic adjuvant-induced arthritis and collagen-induced arthritis models are unable to investigate the pathological role of a single gene [30]. In this study, we applied adenovirus infection methods through the injection of the virus-carrying mutant human gene SAA1.2 to the bone cartridge of the right leg of mice, causing direct gene integration into the mouse genome, and the dosage designation was according to the reported literature [31,32]. Here, we report an optimized experimental protocol completed over a sixweek time course, with optimal times of 2–4 weeks. This investigation strategy can more truly reflect the pathogenic function of a gene during RA initiation and progression and can provide evidence of the negative feedback mechanism of this model. In week 6, the target gene SAA1 was still highly translated, which could be due to the reacted feedback response of articular inflammation, further enhancing the activity of inflammation [28].
Synovitis occurs as a consequence of leukocyte infiltration into the synovium. The accumulation of leukocytes in the synovium does not result from local cellular proliferation but rather from the migration of leukocytes from distant sites of formation in response to the expression of adhesion molecules and chemokines by activated endothelial cells of synovial microvessels [33]. In general, there is a time lag in immune responses in the blood compared with that in the spleen, suggesting that T cell differentiation first occurs in the spleen, and active T cells then circulate in the blood. The synovitis, swelling, and joint damage that characterize active RA are the results of complex autoimmune and inflammatory processes that involve components of both the innate and adaptive immune systems [34]. The innate immune system involves the interaction of fibroblast-like synovial-cytes with other cells of the innate immune system. The intra-articular cellular infiltrates located in the synovial membrane include granulocytes, monocytes/macrophages, natural killer (NK) cells, B cells, and especially CD4+ and CD8+ T cells, all leading to the production of large amounts of chemokines and pro-inflammatory cytokines [19,21,35]. In RA, there is abundant evidence that the innate immune system is persistently activated [36]. In this study, we observed the up-regulated expression of inflammatory cytokines, such as TNF-α, IL-6, IL-22, and G-CSF, which leads to intraarticular immune activity and aggravates joints inflammation. Robust production of IL-22 induces SAA1 production and inflammation [20], and increased TNF-α or IL-17 could enhance MMP production, such as of MMP-3 and MMP-9, leading to articular bone degradation, eventually damaging the joint structure [23–25].
Previously, we have applied molecular docking to compare the protein structures of wild-type SAA1 and SAA1.2 [11]. The SAA1.2 mutation occurred in α-helix 3, and this mutation resulted in a protein conformation change, with a more compressed and condensed protein structure. This mutant may cause more severe inflammatory induction owing to the higher potential for aggregation. Other SAA1 isoforms only deviate from wild-type SAA1 by one amino acid but also exhibit severe inflammatory conditions, highlighting the importance of discovering new SAA1 isoforms or mutants in patients in the future.
We have determined the roles of SAA1.2, SAA1.3, and SAA1.5 in the pathogenesis of RA, as well as the downstream mechanisms by which inflammation is promoted. While all three have pathologic effects, SAA1.2 induces RA symptoms most aggressively. The progression of RA involves multiple organs. The possible mechanism is that SAA1 protein expression starts in synovial fibroblasts and cartilage in the right knee and then circulates in the bloodstream and arrives in the spleen, triggering naive T cell differentiation and mature T cell activation. We found that active cytotoxic CD8+ T cells circulated in the blood and secreted multiple proinflammatory cytokines, resulting in bone destruction (Fig. 8). Collectively, this study highlights the important role of SAA1 isoforms in the initiation and progression of RA, and new drugs targeting SAA1.2 may become novel therapeutics for RA in the future.
《Fig. 8》

Fig. 8. The schematic diagram of arthritis induction by SAA1 isoforms. After intra-articular injection into the knee joint, SAA1 is first expressed by synovial cells and is presented to naive T cells by antigen-presenting cells. Then, T cells become activated and differentiate into Th1, Th17, Th22, or CD8+ T cells, resulting in the release of cytokines like IL-6 and IL-22, which can activate macrophages. In addition, adenoviral-SAA1 transfects into macrophages and induces SAA1 secretion into the synovium, thus activating immune cells to secrete proinflammatory cytokines, such as TNF-α, IL-6, IL-17, and IL-22. Under these conditions, fibroblasts that express receptor activators of TNF-α, IL-1β, or IL-6 can activate macrophages to differentiate via preosteoclasts into osteoclasts that resorb bone from the synovium. These cytokines also activate chondrocytes to secrete MMPs that degrade cartilage. This process starts at the junction between cartilage and bone. Moreover, adenoviral-SAA1 transfects into the blood vessels of the synovium, which promotes endothelial cell secretion of SAA1 and consequently activates T cells in the spleen. Finally, T cell activation and differentiation can produce lymphokines that further promote inflammation in the knee joints via the circulation. IFN: interferon。
《Authors’ contribution》
Authors’ contribution
Liang Liu and Elaine Laihan Leung conceived the research and led the project. Elaine Laihan Leung, Huan-Ling Lai, and Run-Ze Li wrote the manuscript. Liang Liu and Elaine Laihan Leung revised and final proof the manuscript. Huan-Ling Lai, Run-Ze Li, Hu-Dan Pan, Ze-Bo Jiang, Ying Li, Fu-Gang Duan, Jia-Hui Xu, Yi-Zhong Zhang, A-Xi Shi, Chun-Li Wei, Fang-Yuan Zhang, and Xiao-Jun Yao carried out the experiments and analyzed the data. All authors reviewed the manuscript.
《Acknowledgements》
Acknowledgements
This work was funded by The Science and Technology Development Fund, Macao SAR (FDCT-FDCT-17-002-SKL). We thank Dr. David C. Ward who provided insight and expertise that greatly assisted the research.
《Compliance with ethics guidelines》
Compliance with ethics guidelines
Elaine Laihan Leung, Huan-Ling Lai, Run-Ze Li, Hu-Dan Pan, ZeBo Jiang, Ying Li, Fu-Gang Duan, Jia-Hui Xu, Yi-Zhong Zhang, A-Xi Shi, Chun-Li Wei, Fang-Yuan Zhang, Xiao-Jun Yao, and Liang Liu declare that they have no conflict of interest or financial conflicts to disclose.
《Appendix A. Supplementary data》
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.eng.2020.08.018.











 京公网安备 11010502051620号
京公网安备 11010502051620号




