
《1. Introduction》
1. Introduction
Infections caused by multi-drug-resistant (MDR) bacteria are difficult to treat and are becoming commonplace in many institutions. Although attention has previously focused on antibioticresistant Gram-positive organisms, it is now focused on MDR Gram-negative bacteria [1]. The prevalence of these bacteria in clinical settings has increased due to the misuse and overuse of antibiotics and is becoming a global health crisis. According to the China Antimicrobial Surveillance Network (CHINET), in 2018 [2], approximately 30% of resistant clinical isolates were Grampositive bacteria, and the other 70% were Gram-negative bacteria. Among the Gram-negative bacteria, Escherichia coli (E. coli), Klebsiella pneumoniae (K. pneumoniae), and Acinetobacter baumannii (A. baumannii) were the most common. These three bacteria are the main opportunistic pathogens of nosocomial infections, often causing urinary-tract, surgical-site, and pulmonary infections, as well as bacteremia and septicemia. Currently, the resistance of these three Gram-negative bacteria, which have been isolated from clinical patients, to commonly used antibiotics is rising. Continuous surveillance in the Chinese Meropenem Surveillance Study (CMSS) [3] showed that the incidence of carbapenem-resistant E. coli (CREC) from 2010 to 2018 varied between 0.5% and 3.5%, while the incidences of carbapenem-resistant K. pneumoniae (CRKP) and carbapenem-resistant A. baumannii (CRAB) increased from 7.6% to 21.2% and from 64.6% to 69.3%, respectively. CREC, CRKP, and CRAB have been classified by the World Health Organization as belonging to the ‘‘Priority 1: critical” tier of pathogens, indicating the urgent need for the research and development of new antibiotics [4].
The prevalence of MDR Gram-negative bacteria and the lack of development of new antimicrobial agents have prompted the medical community to re-evaluate the use of polymyxin antibiotics. Their use ceased in the 1970s due to nephrotoxicity and neurotoxicity concerns, except as growth promoters in animal husbandry. Polymyxins remain a crucial last-line treatment for infections caused by MDR Gram-negative bacteria and, over the past decade, they have shown effective antibacterial activities against most MDR Gram-negative bacteria in vitro. A worldwide antimicrobial surveillance program (SENTRY) reported low resistance to polymyxins among Gram-negative pathogens between 2006 and 2009 [5]. However, polymyxin-resistant bacteria are being increasingly reported globally in tandem with the frequent use of polymyxins in many institutions [6,7]. The highest resistance rates to polymyxins have been reported primarily for CRKP in Italy (43.0% in 2013–2014) [8], Spain (22.8% in 2010–2012) [9], and Greece (21.7% in 2010–2013) [10]. The prevalence of polymyxinresistant A. baumannii has also increased among CRAB isolates. A microbial resistance surveillance program (EARS-Net) found an overall polymyxin resistance rate of 5% among CRAB isolates from 17 European countries, of which more than 80% were isolated in Greece and Italy [11]. Although the reported polymyxin resistance rate of E. coli is not high, the carriage of the mcr-1 gene in E. coli isolated from animal samples has increased from 5.2% in 2009 to 30.0% in 2014 [12], and there is potential for mammals and birds to act as reservoirs of polymyxin resistance that may be transmitted to humans.
Although the mechanisms underlying polymyxin resistance are not fully understood, several molecular mechanisms have been identified. The majority of polymyxin resistance mechanisms are modifications of lipopolysaccharide (LPS) via the addition of cationic groups to lipid A, and the genes encoding polymyxin resistance are carried on the chromosome or plasmids. LPS also forms an outer membrane (OM) structure that is an important virulence factor in Gram-negative bacteria, potentially inducing strong immune responses in animals [13]. Therefore, mutations that result in the modification of LPS may regulate bacterial fitness and virulence. Fitness cost and compensatory mutations are key to the spread of antimicrobial-resistant bacteria. However, the influences of polymyxin resistance acquisition on the fitness and virulence of these Gram-negative bacteria remain poorly understood. The recent emergence of polymyxin resistance highlights the necessity for an increased understanding of the relationship between different resistance mechanisms, fitness cost, and virulence among these Gram-negative bacteria.
Some studies have evaluated the fitness and virulence of polymyxin-resistant strains. The purpose of this review is to discuss the impact of polymyxin resistance mechanisms on clinically important Gram-negative bacteria (A. baumannii, K. pneumoniae, and E. coli), to investigate the effects of specific mutations on fitness cost and virulence in specific strains, and to predict the clinical outcome for infected patients.
《2. Clinical outcomes of patients with polymyxin-resistant bacteria infections》
2. Clinical outcomes of patients with polymyxin-resistant bacteria infections
For the treatment of severe MDR Gram-negative bacterial infections, polymyxins are considered to be the most effective antibiotics; therefore, polymyxin resistance is a severe hindrance in the treatment of such infections. A retrospective study undertaken at Singapore General Hospital found that the development of polymyxin resistance in carbapenem-resistant Enterobacteriaceae (CRE) was associated with poor clinical outcomes [14]. It showed that patients in the polymyxin-resistant CRE group suffered from a higher 30-day mortality rate (50%), longer intensive care unit stay, and higher occurrence of co-infections than patients in the polymyxin-susceptible CRE group. Similarly, Qureshi et al. [15] found that the 30-day all-cause mortality rate was 30% in 20 patients infected with polymyxin-resistant CRAB. The emergence of polymyxin-resistant CRE and CRAB is problematic because there are no effective antibiotics to treat infections caused by them. Qureshi et al. [15] also pointed out that a history of recent polymyxin exposure was a significant risk factor because 19 out of 20 patients infected with polymyxin-resistant CRAB had previously used polymyxin. Although it is not described how polymyxin was administered (e.g., dosing, course of treatment, and monotherapy or combinational therapy), the improper use of these antibiotics may promote the development of drug resistance in pathogens. Therefore, more attention should be paid to polymyxin resistance. In 2019, six international academic organizations reported consensus guidelines for the optimal use of polymyxins [16]. These practical guidelines provide detailed therapeutic recommendations regarding polymyxin agent selection, dosing, the use of monotherapy or combinational therapy, and special drug regimens for patients with hepatic or renal dysfunction. It is noted that the usage of polymyxins should be strictly regulated to avoid the rapid development of resistance to them. Polymyxins should not be used in the decolonization of asymptomatic CRE carriers [17] or in selective decontamination of the digestive tract for prophylaxis medication [18].
《3. The mode of action of polymyxins》
3. The mode of action of polymyxins
In Gram-negative bacteria, the OM is a permeable and protective barrier to external attacks, including various antibiotics [19,20]. Polymyxins can directly combine with polyanionic lipid A of LPS [21] to disrupt the structure and function of the OM. The specific molecular mechanism is as follows. Polymyxins bind to LPS through an initial electrostatic interaction with the α, -diaminobutyric acid in lipid A, thereby displacing the divalent cations Ca2+ and Mg2+ from LPS [19,22]. The polymyxin molecule then inserts its hydrophobic domains into the fatty acyl chain of LPS and subsequently into the inner membrane (IM) leaflet [23–25], destabilizing the structure and function of the bacterial membrane, which leads to leakage of the cytoplasmatic contents and, finally, bacterial death [26].
-diaminobutyric acid in lipid A, thereby displacing the divalent cations Ca2+ and Mg2+ from LPS [19,22]. The polymyxin molecule then inserts its hydrophobic domains into the fatty acyl chain of LPS and subsequently into the inner membrane (IM) leaflet [23–25], destabilizing the structure and function of the bacterial membrane, which leads to leakage of the cytoplasmatic contents and, finally, bacterial death [26].
Another possible target involved in the anti-bactericidal activity of polymyxins is an essential respiratory enzyme (type II nicotinamide adenine dinucleotide (NADH)-quinone oxidoreductase) in the cytoplasmic membrane [27]. Several studies have shown that polymyxins can inhibit bacterial respiration via the inhibition of type II NADH-quinone oxidoreductase activity in three Gramnegative species (K. pneumoniae, E. coli, and A. baumannii) in a concentration-dependent manner [28–30].
《4. Mechanisms of polymyxin resistance in Gram-negative bacteria》
4. Mechanisms of polymyxin resistance in Gram-negative bacteria
《4.1. Modification of LPS mediated by genes encoded on the chromosome and plasmids》
4.1. Modification of LPS mediated by genes encoded on the chromosome and plasmids
Acquired resistance to polymyxin has been identified in K. pneumoniae, E. coli, and A. baumannii, and is predominantly due to modification of LPS by the addition of phosphoethanolamine (pEtN) and/or 4-amino-4-deoxy-L-arabinose (L-Ara4N) cationic groups to lipid A. Modifications of LPS are mainly regulated by the polymyxin resistance gene A/B-coded protein (PmrA/PmrB) and phosphate regulon P/Q (PhoP/PhoQ) two-component systems. The specific mechanism is as follows (Fig. 1). The synthesis of pEtN and/or its addition to LPS is mediated by the pmrCAB operon, whereas the synthesis of L-Ara4N and/or its addition to LPS is mediated by the pmrHFIJKLM operon. In the PmrA/PmrB twocomponent system, PmrB, a sensor tyrosine kinase located in the IM, can phosphorylate PmrA, which is a responsory regulator of PmrB [31,32]. Phosphorylated PmrA binds to the pmrCAB and pmrHFIJKLM operon promoter regions and successively activates their transcription, leading to the synthesis of the cationic groups pEtN and L-Ara4N, and their addition to lipid A [32,33].
《Fig. 1》
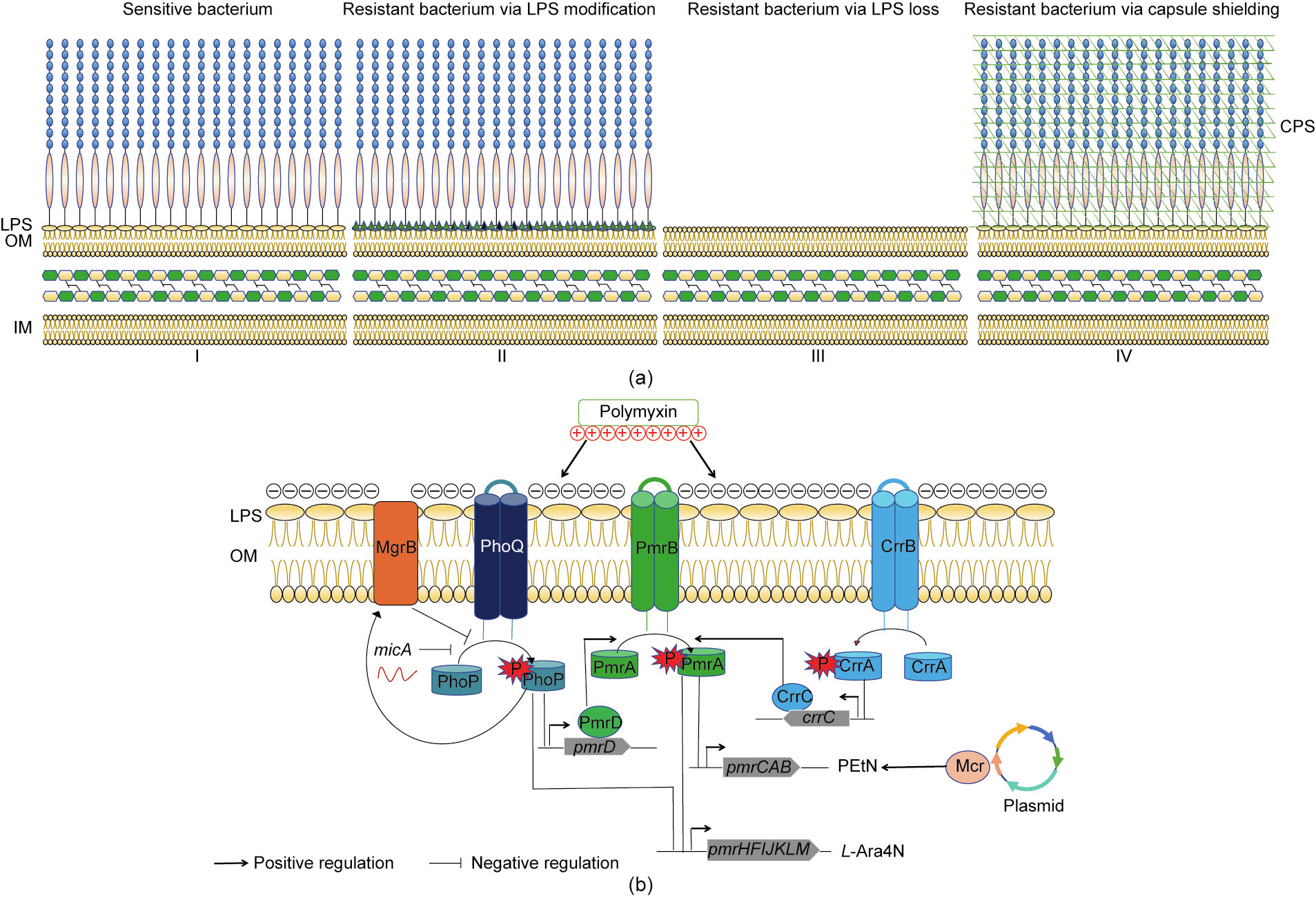
Fig. 1. (a) Different mechanisms of polymyxin resistance in three Gram-negative bacteria. I. Components of the cell membrane of a sensitive Gram-negative bacterium. II. E. coli, K. pneumoniae, and A. baumannii can acquire polymyxin resistance by LPS modification (green triangles represent the cationic groups). III. In A. baumannii, complete loss of LPS can lead to polymyxin resistance. IV. K. pneumoniae can become resistant due to overexpression of capsular polysaccharide (CPS). (b) LPS modifications mediated by genes encoded on the chromosome and plasmids. P: a phosphate group; MgrB: Mg2+-responsive gene B-coded protein; CrrA/B/C: colistin resistance regulation gene A/B/Ccoded protein; Mcr: mobile colistin resistance gene-coded protein.
Chemical modification of lipid A can also be regulated by another two-component system, PhoP/PhoQ [34–36]. In the same way as PmrA/PmrB, PhoQ, a sensor tyrosine kinase located in the IM, can phosphorylate PhoP, which is a responsory regulator of PhoQ [34,35]. Phosphorylated PhoP can directly bind to and activate the transcription of the pmrHFIJKLM operon, which subsequently adds L-Ara4N to LPS [32]. The PhoP/PhoQ signaling pathway can also mediate lipid A modification via genes in the PmrA/PmrB signaling pathway. Furthermore, phosphorylated PhoP can indirectly activate the PmrA/PmrB two-component system through the PmrD connector protein, which protects PmrA from dephosphorylation [37].
In K. pneumoniae, the PmrA/PmrB and PhoP/PhoQ twocomponent systems are further regulated by the CrrA/CrrB twocomponent system [38] and mgrB [39–44], respectively. The physiological function of CrrA/CrrB has not been fully clarified. Inactivation of CrrB can promote the expression of the pmrHFIJKLM and pmrCAB operons [38]. MgrB, a small transmembrane protein [45], plays a key role in negative feedback regulation of the PhoP/PhoQ two-component system. The mgrB gene is upregulated following the phosphorylation of PhoP. In turn, the MgrB protein inhibits the expression of phoQ, resulting in a decrease in phosphorylated PhoP [45]. Inactivation of mgrB promotes the overexpression of the PhoP protein, leading to high levels of lipid A modifications.
In addition to the abovementioned chromosomal genes, mcr, a newly identified gene that is predominantly harbored on diverse plasmids, is involved in polymyxin resistance. This gene encodes pEtN transferase, the expression of which leads to the addition of pEtN to lipid A. In 2015, Liu et al. [46] were the first to identify the plasmid-borne mobilized colistin resistance gene mcr-1. Since then, many studies have found other mcr alleles, including mcr2–9 [47–51]. The mcr genes have been found in several species of Enterobacteriaceae, such as E. coli [46], K. pneumoniae [46], and Salmonella enterica [52].
《4.2. Loss of LPS mediated by the lpx gene》
4.2. Loss of LPS mediated by the lpx gene
Another mechanism of polymyxin resistance observed in A. baumannii is the complete loss of LPS on the cell surface due to mutations in the lipid A synthesis genes. LPS is synthesized by the lipopolysaccharide peroxidation (LpX) pathway [53,54] (including lpxA, lpxC, and lpxD) in the cytoplasm and transported to the OM by the lipopolysaccharide transport (LpT) pathway [55]. Mutations in lpxA, lpxC, and lpxD caused by substitutions, frameshifts, truncations, or insertional inactivation result in polymyxin resistance. lpx-mediated polymyxin resistance has only been observed in A. baumannii.
《4.3. Overexpression of capsular polysaccharide》
4.3. Overexpression of capsular polysaccharide
Another intriguing mechanism leading to polymyxin resistance in K. pneumoniae is the overproduction of capsular polysaccharide (CPS), which is attached to the cell surface through an electrostatic interaction with LPS [56]. One study showed that CPS limits the interaction of polymyxins and LPS in K. pneumoniae and that purified CPS binds directly to polymyxins [56].
In conclusion, E. coli and K. pneumoniae can acquire polymyxin resistance via LPS modification mediated by chromosomal genes (pmrA/pmrB and phoP/phoQ) and plasmid-mediated polymyxin resistance genes (mcr). In addition, K. pneumoniae can acquire polymyxin resistance via crrA/crrB or mgrB mutations or from the overexpression of CPS. The plasmid-mediated polymyxin resistance gene mcr-1 has been found in E. coli and K. pneumoniae, but not in A. baumannii. Nonetheless, A. baumannii can acquire polymyxin resistance through the complete loss of LPS caused by mutations in the lipid A synthesis genes (Fig. 1).
《5. Effects of different polymyxin resistance mechanisms on virulence and fitness》
5. Effects of different polymyxin resistance mechanisms on virulence and fitness
Virulence is defined as ‘‘the relative capacity of a microorganism to cause damage in a host” [57]. Whether pathogens can cause diseases in the host depends on the balance between bacterial virulence and host immunity. Bacterial virulence factors can be divided into exotoxins and endotoxins according to their sources, properties, and functions. The lipid A component of LPS plays a critical role in immune and inflammatory responses through the initial release of cytokines (tumor necrosis factor-α (TNF-α) and interleukin (IL)-8) [58]. As such, LPS in Gram-negative bacteria is considered to be an endotoxin [59]. In addition to LPS, other virulence factors that have been observed in K. pneumoniae include CPS, siderophores, and fim-briae [60]. E. coli can also display a variety of virulence factors, such as fimbriae, flagella, non-fimbriae adhesin, α-hemolysin, cytolethal tumefaction toxin, an iron acquisition system, a capsule, and OM protein A (OmpA) [61]. A. baumannii has complex virulence factors [62], including LPS [63,64], biofilm, OmpA, exopolysaccharide and capsule formation [65], efflux pumps, and penicillin-binding proteins.
Resistance is often related to a decline in bacterial fitness [66], and an overuse of antibiotics exerts a strong selective pressure on bacteria to acquire resistance. Sensitive bacteria generally outcompete resistant bacteria in the absence of antibiotics due to their higher fitness [66]. However, through compensatory mutations, genetic co-selection, and unknown factors, resistant bacteria can negate the biological cost and stably exist in the bacterial population [66].
Several studies have identified alterations in virulence and fitness in polymyxin-resistant Gram-negative bacteria. In some studies, polymyxin-resistant strains were obtained directly from patients during treatment with polymyxin, while in others, clinically derived sensitive strains or laboratory standard strains were exposed to increased concentrations of polymyxin in order to induce the production of resistant isolates in vitro. The fitness and virulence of both polymyxin-resistant and polymyxinsensitive bacteria were then assessed.
The mortality of the host or the reproduction rate of bacteria in the host was used to measure virulence [67]. Three animal models were established to evaluate bacterial virulence, including Caenorhabditis elegans, Galleria mellonella (G. mellonella), and mouse infection models. The fitness cost of resistance is usually evaluated by measuring the exponential growth rates of sensitive and resistant bacteria in vitro and by calculating a competition index in vivo and in vitro (Fig. 2) [66]. The effects of different polymyxin resistance mechanisms on virulence and fitness in A. baumannii, K. pneumoniae, and E. coli are listed in Table S1 in Appendix A.
《Fig. 2》
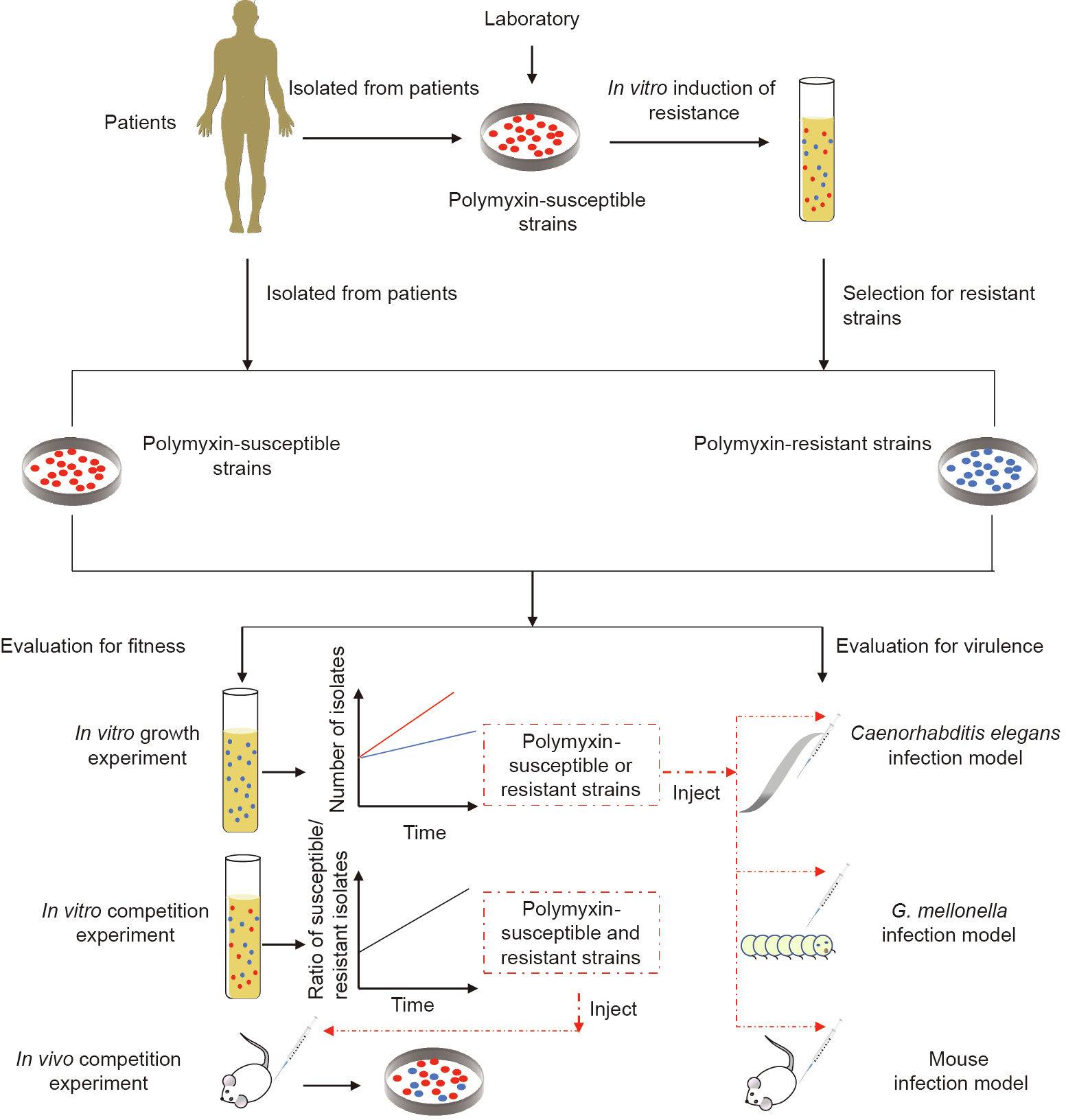
Fig. 2. General experimental flow scheme. Polymyxin-resistant strains were isolated from clinical patients or obtained via the in vitro induction of resistance in susceptible strains. Virulence is usually evaluated by three animal models: Caenorhabditis elegans, G. mellonella, and mouse infection models. The fitness cost of the strains is usually evaluated by measuring the exponential growth rates in vitro and the competition index in vivo and in vitro.
《5.1. A. baumannii》
5.1. A. baumannii
5.1.1. Effects of pmrA and pmrB mutations on virulence and fitness
Polymyxin resistance caused by pmrA and pmrB mutations in clinical A. baumannii often leads to reduced virulence and fitness. Hraiech et al. [68] studied two strains of A. baumannii isolated from a patient with pneumonia; one was susceptible to polymyxin and the other was resistant. Polymyxin resistance was caused by mutations in pmrA (E8D) and the loss of a prophage [68]. The resistant strain grew more slowly than the susceptible strain in vitro [68]. In a rat pneumoniae infection model, rats infected by the resistant strain showed milder symptoms, such as lower bacterial counts, more confined systemic dissemination, less severe lung injury, and a better outcome, than those infected by the susceptible strain [68]. Clinical A. baumannii isolates that have acquired polymyxin resistance through pmrA mutations, including single amino acid substitutions such as M12K [69], D82G [70], and S119T [70], consistently exhibit impaired virulence and decreased fitness in vivo and in vitro [69,70]. During the development of resistance in vivo, Jones et al. [71] found that early-isolated polymyxin-resistant strains in the same patient were outcompeted by late-isolated polymyxin-resistant strains. However, no single nucleotide polymorphisms were detected in the virulence genes of the polymyxin-resistant strains [71]. Compensation of virulence loss in late-stage resistant isolates may be due to post-translational modifications or physiological changes [71].
A. baumannii isolates from severe clinical cases treated with polymyxin acquired polymyxin resistance via various pmrB mutations [70–78], including the single amino acid substitutions P233S, P170L, G21V, V227A, I232T, A28V, and S17R, and the deletions 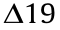 and –G12. Most mutations were associated with fitness cost and impaired virulence, but there were some contradictory results with respect to specific pmrB mutations (P233S and P170L). Two A. baumannii isolates, Ab249 and Ab347, carried pmrB P233S and P170L mutations, respectively. Both strains exhibited reduced in vitro exponential growth rates and reduced in vitro and in vivo virulence [72,73]. Both polymyxin-resistant A. baumannii strains under-expressed proteins with important functions in biofilm formation (CsuA/B and C) and the oxidative stress response (aconitase B, KatE catalase, superoxide dismutase, and alkyl hydroperoxide reductase) [72]. The decreased virulence may be related to the reduction of initial cell adhesion and consequent reduction of biofilm formation [73]. However, Leite et al. [70] showed that polymyxin-resistant A. baumannii carrying the pmrB P170L mutation was more virulent than the sensitive strain toward G. mellonella. This polymyxin-resistant isolate belonged to a different sequence type (ST) 233, which was not one that circulated in the studied hospital. However, it should be noted that Leite et al. [70] studied A. baumannii isolated from different patients; therefore, the genetic background of the strain may be different.
and –G12. Most mutations were associated with fitness cost and impaired virulence, but there were some contradictory results with respect to specific pmrB mutations (P233S and P170L). Two A. baumannii isolates, Ab249 and Ab347, carried pmrB P233S and P170L mutations, respectively. Both strains exhibited reduced in vitro exponential growth rates and reduced in vitro and in vivo virulence [72,73]. Both polymyxin-resistant A. baumannii strains under-expressed proteins with important functions in biofilm formation (CsuA/B and C) and the oxidative stress response (aconitase B, KatE catalase, superoxide dismutase, and alkyl hydroperoxide reductase) [72]. The decreased virulence may be related to the reduction of initial cell adhesion and consequent reduction of biofilm formation [73]. However, Leite et al. [70] showed that polymyxin-resistant A. baumannii carrying the pmrB P170L mutation was more virulent than the sensitive strain toward G. mellonella. This polymyxin-resistant isolate belonged to a different sequence type (ST) 233, which was not one that circulated in the studied hospital. However, it should be noted that Leite et al. [70] studied A. baumannii isolated from different patients; therefore, the genetic background of the strain may be different.
Durante-Mangoni et al. [75] reported a resistant clinical strain with a pmrB P233S mutation, but the mutation was not related to a loss of virulence in the G. mellonella infection model or a decreased in vitro growth rate. Wand et al. [79] found that mutations in pmrB P233S do not always lead to a decrease in virulence. Twelve polymyxin-susceptible clinical A. baumannii isolates acquired polymyxin resistance following the in vitro stepwise selection of polymyxin-resistant isolates. Among them, the virulence of two resistant strains with 17–26 duplication and T235I mutations in pmrB was similar to that of their corresponding parental strains in the G. mellonella infection model [79]. Other resistant strains with pmrB mutations show weakened virulence and adaptability [79], and the polymyxin-resistant derivatives of A. baumannii with pmrB mutations A227V [80], N353Y [80], S17R [79], R134C [81], and G272D [82] showed reduced in vitro and in vivo fitness and virulence.
5.1.2. Effects of lpxA, lpxC, and lpxD mutations on virulence and fitness
Published studies [79,80,82–85] all support the contention that mutations of lpx genes are associated with impaired virulence and a fitness cost. In one study, Carretero-Ledesma et al. [83] found that LPS-deficient strains caused by lpxA, lpxC, and lpxD mutations induced lower TNF-α and IL-6 serum levels than the corresponding clinical parental polymyxin-sensitive strain and led to lower mortality in a mouse systemic infection model. Loss of LPS influences biofilm production and surface motility [83]. In another study, Wand et al. [79] found that clinical polymyxin-resistant A. baumannii isolates caused by single mutations in lpxA (E216*), lpxC (I253N, F191L, and A82E), or lpxD (K318frameshift), or by the inactivation of lpxC (lpxC::ISAba1), were correlated with decreased growth rates and virulence in G. mellonella. They also compared the effects of mutations in pmrB and lpx on fitness and virulence; the results showed that mutations in lpx had a more important role in the alteration of fitness and virulence [79]. In addition to pmr and lpx, a mutation in the pmrC homologue eptA and a point mutation in ISAba1 upstream of eptA were studied in clinical polymyxin resistant A. baumannii [76]. Fitness and virulence were not decreased in these isolates. However, there is limited understanding of the relationship between eptA and polymyxin resistance.
《5.2. K. pneumoniae and E. coli》
5.2. K. pneumoniae and E. coli
5.2.1. Effects of pmr and pho mutations on virulence and fitness
Few studies report on the fitness and virulence of polymyxinresistant K. pneumoniae resulting from mutations in pmrA/pmrB or phoP/phoQ. In one study, mutations were observed in pmrA (G53C), pmrB (229–261 duplication, P95L, G53C, 213–261 duplication,  150Y, D51–59, and T157P), and phoQ (L348Q and T244N)[86]. However, there was no clear association between the sites of specific polymyxin resistance mutations and the variation of virulence observed in the G. mellonella infection model [86]. Rather, the retention of fitness appeared to be influenced more by specific strain backgrounds, with some strains being capable of accommodating different resistance mutations with no significant loss of virulence. However, decreased virulence and fitness were observed in another study [87]. These polymyxin-resistant K. pneumoniae isolates showed reduced CPS production, serum resistance, biofilm formation, and growth rates [87]. Lipid A was found to be modified by the addition of L-Ara4N and palmitate in polymyxin-resistant strains when analyzed by matrix-assisted laser desorption/ ionization-time of flight (MALDI-TOF) mass spectrometry [87].
150Y, D51–59, and T157P), and phoQ (L348Q and T244N)[86]. However, there was no clear association between the sites of specific polymyxin resistance mutations and the variation of virulence observed in the G. mellonella infection model [86]. Rather, the retention of fitness appeared to be influenced more by specific strain backgrounds, with some strains being capable of accommodating different resistance mutations with no significant loss of virulence. However, decreased virulence and fitness were observed in another study [87]. These polymyxin-resistant K. pneumoniae isolates showed reduced CPS production, serum resistance, biofilm formation, and growth rates [87]. Lipid A was found to be modified by the addition of L-Ara4N and palmitate in polymyxin-resistant strains when analyzed by matrix-assisted laser desorption/ ionization-time of flight (MALDI-TOF) mass spectrometry [87].
5.2.2. Effects of mgrB mutations on virulence and fitness
Arena et al. [88] used a G. mellonella infection model to compare the virulence of two ST258 K. pneumoniae carbapenemase (KPC)- producing K. pneumoniae isolates with isogenic polymyxinresistant mgrB mutants produced by insertional inactivation. The mgrB mutants showed a similar virulence level as the parental strain. Another study also found that fitness did not significantly differ between polymyxin-sensitive and -resistant strains caused by the insertional inactivation of mgrB when assessed by in vitro competition experiments [89]. Kidd et al. [90] revealed that mgrB mutations induce phoP/phoQ-regulated lipid A remodeling, which not only results in polymyxin resistance, but also promotes virulence of K. pneumoniae by suppressing activation of the early host defense response [90]. Overall, inactivation of mgrB does not lead to significantly decreased virulence or a fitness cost.
5.2.3. Effects of mcr-1 mutation on virulence and fitness
Tietgen et al. [91] transferred an expression vector carrying mcr-1 into E. coli J53 and K. pneumoniae PRZ transformants. Competition experiments showed equal growth rates between E. coli J53 transconjugants and the parental strain, while K. pneumoniae PRZ transconjugants exhibited lower growth rates than the parental strain. Virulence was not changed for mcr-1 transformants (E. coli J53 transconjugants and K. pneumoniae PRZ transconjugants) assayed in A549 human lung epithelial cells and the G. mellonella infection model [91]. The fitness cost caused by the acquisition of mcr-1 was observed in E. coli, but not in K. pneumoniae. This is consistent with the phenomenon that mcr-1 is found more frequently in E. coli than in K. pneumoniae. In vitro and in vivo growth rates were decreased in a mcr-1-positive K. pneumoniae strain in another study [92].
《6. Biological costs of drug resistance for other antimicrobial peptides》
6. Biological costs of drug resistance for other antimicrobial peptides
Bacitracin, an important peptide antibiotic, is produced mainly by Bacillus licheniformis and Bacillus subtilis [93]. Bacitracin and polymyxin have been widely used as growth promoters in livestock animals [94]. Bacitracin disrupts the synthesis of the cell wall of most Gram-positive and some Gram-negative bacteria by inhibiting dephosphorylation, eventually leading to leakage of cellular contents and cell death. However, unlike polymyxin, resistance to bacitracin always seems to be associated with higher fitness and virulence. One study reported the function of a novel membrane transporter module SstFEG in bacitracin resistance, which functions not only as an efflux pump for bacitracin resistance, but also as a virulence-related protein in Streptococcus suis [95]. Bacitracin resistance was associated with increased fitness in Lactococcus lactis (L. lactis), as determined by an accelerated growth rate and doubled biomass production. The researchers used this phenomenon to improve the large-scale production of L. lactis and to obtain the required secretion of nisin A, a highly efficient and safe preservative for food products [96]. This is a good example of using the association of resistance with increased fitness to develop industries.
《7. Conclusions》
7. Conclusions
For A. baumannii, most of the literature focuses on the impact of polymyxin resistance caused by pmr, pho, and lpx on fitness cost and virulence. Most studies reported decreased virulence and a fitness cost in polymyxin-resistant A. baumannii. Moreover, mutation of lpx leading to complete loss of LPS has a greater influence on fitness and virulence than mutations of pmr and pho leading to modification of LPS [79]. However, the same mutation may have different effects on fitness cost and virulence; examples include pmrB P170L and P233S mutations, which decrease fitness and virulence in some studies [72,73,79] while in others the opposite is the case [70,75,79]. Compensation for biological cost may result from the presence of compensatory mutations in other genes. Overall, the fitness cost caused by polymyxin resistance would help to limit the transmission of polymyxin-resistant A. baumannii in a clinical environment. This phenomenon is consistent with the observed sporadic infections of polymyxin-resistant A. baumannii.
For K. pneumoniae,most studies focus on the impact of polymyxin resistance on fitness cost and virulence caused by pmr, pho, mgrB, and mcr-1 mutations. Most studies report decreased virulence and a fitness cost in polymyxin-resistant strains with pmr and pho mutations, with no change in polymyxin-resistant strains with mgrB mutations. The fitness cost caused by the acquisition of mcr-1 was found to depend on species, applying not to E. coli but to K. pneumoniae [91]. However, some studies have shown that acquiring mcr-1 can reduce virulence [92], while some researchers think that acquiring mcr-1 does not affect virulence [91].
Compared with A. baumannii, there is little direct correlation between mutations in particular genes linked to polymyxin resistance and the retention of fitness/virulence in K. pneumoniae [86]. The impact of virulence and the biological cost in K. pneumoniae is more likely to be related to the genetic background of the test strains. Clinical strains, especially those isolated from different patients, have different genetic backgrounds. For example, strains isolated from different patients that are used for comparison may have significant differences in baseline virulence and fitness levels due to the carriage of different virulence genes and grouping into different STs.
For E. coli, there are few studies concerning the impact of polymyxin resistance mechanisms on virulence and fitness cost. Studies have mainly examined the impact of the acquisition of mcr-1 on virulence and fitness cost [91], and such acquisition has not been found to change virulence and fitness [91].
In conclusion, the biological cost of drug-resistant bacteria may be the key to reversing resistance. Most polymyxin resistance mechanisms are associated with impaired fitness and virulence, leading to decreased competitiveness of the resistant strains compared with susceptible strains in the absence of antibiotics. However, not all polymyxin-resistant strains showed decreased fitness and virulence; some were as virulent or more virulent than their counterpart susceptible strains, and did not exhibit an increased fitness cost. Thus, we must be vigilant against the prevalence of these polymyxin-resistant isolates with unchanged or higher virulence and fitness, because they are more likely to survive in a clinical environment and become an important factor in the spread of polymyxin resistance. The reasons for this phenomenon are not completely understood. Compensatory cost-free mutations and genetic co-selection may be obstacles in the elimination of drug-resistant bacteria [66]. Therefore, more in-depth and basic research is needed to fully understand the interaction between drug resistance and bacterial biology, which may help to develop interventions for controlling the spread of polymyxinresistant strains. As for bacitracin, detailed knowledge of the physiological basis of fitness costs could be used to select and design new therapies exploiting the highest cost of resistance and the lowest probability of compensation by adaptive mutations.
《Acknowledgments》
Acknowledgments
This work was supported by the National Key Research and Development Program of China (2017YFC1600100 and 2017YFC1200203), the National Natural Science Foundation of China (81702040), and the National Science Foundation of Zhejiang Province, China (LY20H190002).
《Compliance with ethics guidelines》
Compliance with ethics guidelines
Yuan Wang, Qixia Luo, Tingting Xiao, Yunying Zhu, and Yonghong Xiao declare that they have no conflict of interest or financial conflicts to disclose.
《Appendix A. Supplementary data》
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.eng.2020.11.005.











 京公网安备 11010502051620号
京公网安备 11010502051620号




