

《前 言》
前 言
微生物基因组计划 (Microbial Genome Program, MGP) 及后基因组研究是继人类基因组计划以外生命科学领域启动的又一巨大工程。通过对微生物基因组及功能基因的研究, 不仅能够使人们更深入地了解病原微生物的繁殖代谢、致病和耐药等机理, 寻找更灵敏及特异的用于诊断和分型的分子标记以及有效的药物作用靶标, 而且可为临床筛选有效的药物及发展疫苗提供基础。
志贺菌属 (shigella) 细菌通称痢疾杆菌, 是一类具有高度传染性和危害严重的革兰氏阴性肠道致病菌, 临床感染可以导致痢疾 (shigellosis) , 其症状以发热、脱水和便血为特征。痢疾是世界上, 尤其是发展中国家重要的传染病之一。全球每年的病例超过1.6亿, 并导致110万患者死亡, 其中绝大多数为5岁以下的儿童, 因此痢疾是世界上造成婴幼儿死亡的主要原因之一
福氏2a志贺菌301株 (Shigella flexneri 2a strain 301, Sf301) 全基因组序列测定是我国第一个在国际上率先发布并完成的微生物基因组计划, 所得到的大量重要数据和信息将为相关研究提供重要线索。本文重点介绍痢疾杆菌基因组中的特异性序列以及SIs的结构特征及功能预测, 以期有助于更好地理解痢疾杆菌的进化和致病机理, 为进一步研究痢疾的预防和治疗措施奠定基础。
毒力岛 (Pathogenicity Island, PAIs) 是医学细菌学领域的新名词, 是指一类编码成簇毒力相关基因的、相对分子量较大、其DNA片段的G+C含量及密码子使用情况与宿主细菌染色体明显不同, 且遗传相对不稳定的染色体DNA片段
《1 材料与方法》
1 材料与方法
《1.1菌株和生长条件》
1.1菌株和生长条件
福氏2a志贺菌301株:于1984年从北京市昌平县痢疾患者的粪便标本中分离。该菌株一直被作为中国福氏志贺菌的参考株, 由中国预防医学科学院流行病学研究所提供。
菌株在常规刚果红培养基中37℃培养过夜, 挑取红色菌落在不含抗生素的LB培养基中于37℃振荡培养过夜, 用于分离大质粒和染色体DNA。
《1.2鸟枪法序列测定与拼接》
1.2鸟枪法序列测定与拼接
分别用改良CTAB法
序列拼接应用Phred/Phrap
《1.3基因组注释》
1.3基因组注释
采用Glimmer2.0软件
tRNA 基因的识别采用tRNAscan-SE 程序
《1.4比较基因组分析》
1.4比较基因组分析
与大肠杆菌K-12MG1655 (GenBank登记号为U00096) 的全基因组
《1.5核酸序列登记号》
1.5核酸序列登记号
Sf301的染色体及大质粒pCP301的核苷酸序列已提交GenBank, 登记号分别为AE005674和AF386526。
《2 结果》
2 结果
《2.1基因组基本特征》
2.1基因组基本特征
Sf301株的全基因组主要包括染色体和侵袭性大质粒 (简写为pCP301) 两部分, 其基本特征列于表1。环型染色体全长4 607 203 bp, G+C 含量为0.5089 mol, 包含7套16S-23S-5S rRNA操纵子, 1个tmRNA和包括RNase P, 6S RNA 及 4.5S RNA在内至少9个未分类RNA;染色体全长的80.4%为蛋白编码区, 0.8% 编码稳定的RNA, 而314个完整或截短拷贝的IS元件占到全长的3.6%。预测的开放读码框总数为4 434个, 除去由于碱基插入、缺失、读框内终止以及移码突变而导致的254个假基因外, ORFs的平均读长为891 bp。在所有的4 180个完整ORFs中, 有2 895个 (65%) 可以分类到COGs库的不同生物功能种类, 268个 (6.4%) 只有简单的功能预测, 而另外1 195个 (28.6%) 则只是推断的功能未知的编码序列, 其中有179个ORF与目前已知的任何蛋白质都没有显著的同源性。编码蛋白在COGs库中的功能分类情况列于表2。
表1 Sf301 基因组基本特征 Table 1 General features of Sf301 genome
《表1》
| 染色体 | 大质粒 | |
| 总长 (碱基对) | 4 607 203 | 221 618 |
| 开放读框总数 | 4 434 | 267 |
| 开放读框平均长度 (碱基对) | 891 | 658 |
| 序列编码率/% | 80.4 | 76.24 |
| G+C 含量/% | ||
| 全长 | 50.89 | 45.77 |
| 蛋白编码区 | 51.96 | 46.13 |
| RNA 基因 | 54.79 | - |
| 基因间区域 | 46.07 | 44.59 |
| 插入序列元件 | 314 | 88 |
| 其中不完整拷贝数 | 67 | 62 |
| 核糖体 RNA | ||
| 16S | 7 | - |
| 23S | 7 | - |
| 5S | 8 | - |
| 转运 RNA数目 | 97 | - |
| tmRNA | 1 | - |
| 未分类 RNA数目 | 9 | - |
大质粒pCP301全长221 618 bp, 共编码267个ORFs, 除去6个假基因, 其平均读长为658 bp, 序列编码率为76.24%, 编码区的G+C含量为0.4613 mol。 pCP301中含有68 kb的IS元件, 占到总长度的30%, 包括18个IS元件种类共计111个片段。
《2.2复制起始与终止区》
2.2复制起始与终止区
沿用大肠杆菌K-12 MG1655确定的染色体的第一个碱基, 痢疾杆菌福氏2a 301株的第一个碱基选定在无明显特征的lasT和thrL两基因之间。通过GC偏斜 (GC skew ) [ (G-C) / (G+C) ]分析以及与大肠杆菌K-12 MG1655的相似性, 确定了Sf301染色体复制原点oriC。我们选择gidA 和 mioC基因间Bgl Ⅱ内切酶位点的鸟嘌呤残基作为Sf301染色体复制原点的第一个核苷酸。GC偏斜分析显示, Sf301染色体的复制终止点 (terC) 位于染色体的1.60 Mb 附近。
表2 Sf301染色体蛋白在COGs系统中的分类 Table 2 Distribution of Sf301 chromosomal proteins among COGs functional groups
《表2》
| Functional class (COGs) | Number | percent of total/% |
| 翻译、核糖体结构及生物发生 | 159 | 3.80 |
| 转录 | 185 | 4.43 |
| DNA复制、重组与修复 | 615 | 14.71 |
| 细胞分裂与染色体分离 | 28 | 0.67 |
| 翻译后修饰、蛋白周转、伴侣 | 108 | 2.58 |
| 细胞外套的生物合成、外膜 | 168 | 4.02 |
| 细胞动力和分泌 | 106 | 2.54 |
| 无机离子的转运和代谢 | 153 | 3.66 |
| 信号转导系统 | 89 | 2.13 |
| 能量产生与转化 | 217 | 5.19 |
| 碳水化合物的转运与代谢 | 264 | 6.32 |
| 氨基酸的转运与代谢 | 294 | 7.03 |
| 核苷的转运与代谢 | 74 | 1.77 |
| 辅酶的代谢 | 117 | 2.80 |
| 类脂化合物的代谢 | 68 | 1.63 |
| 次级代谢产物的生物合成, 、转运及分解代谢 | 72 | 1.72 |
| 预测的一般功能 | 268 | 6.41 |
| 功能未知和未分类的 | 1 195 | 28.59 |
| 总数 | 3 180 | 100 |
《2.3痢疾杆菌特异性的DNA片段——SIs的分析》
2.3痢疾杆菌特异性的DNA片段——SIs的分析
Sf301的染色体与大肠杆菌K-12 MG1655株染色体长度非常接近 (4 639 221 bp)
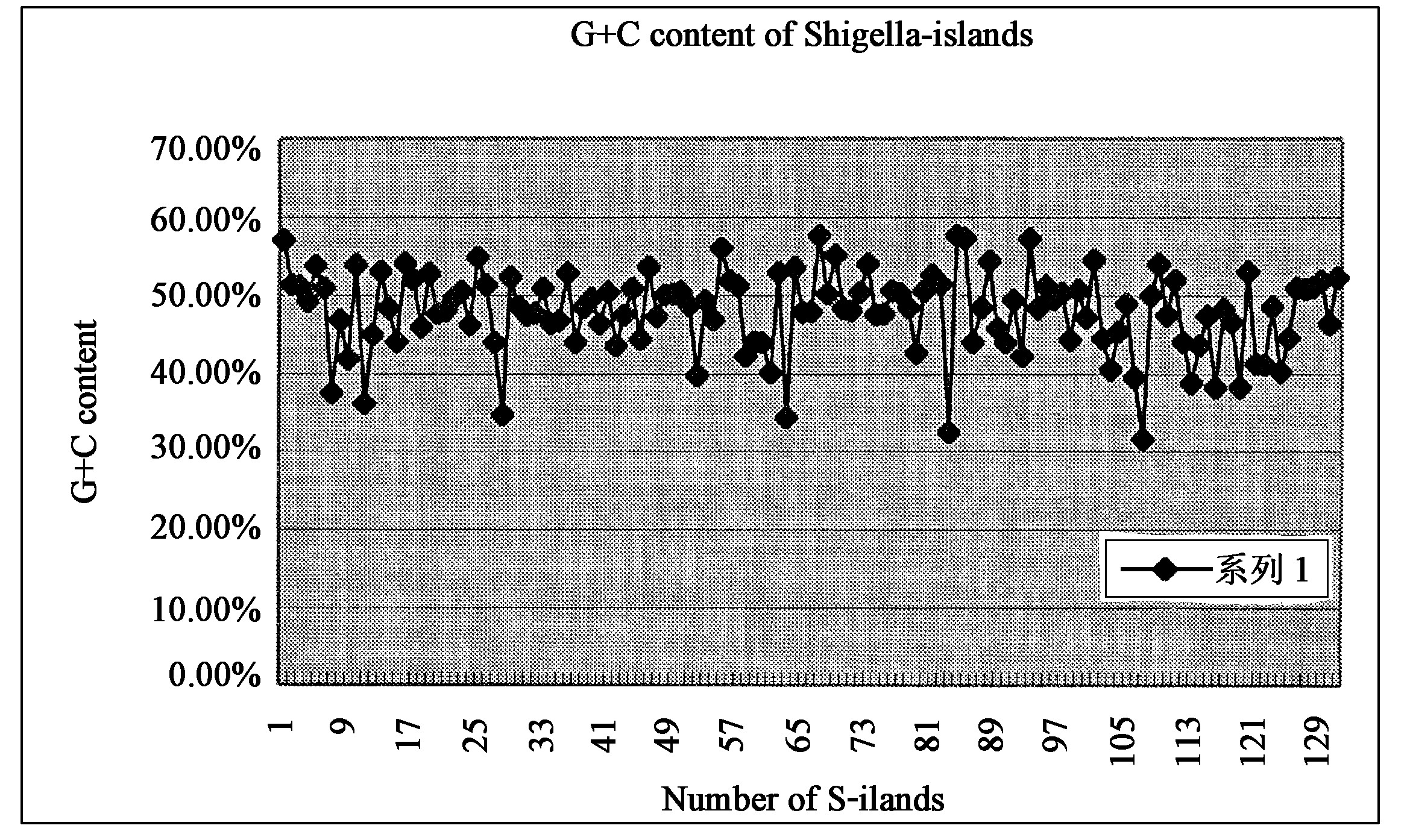
SIs的平均G+C含量为0.4825 mol, 显著低于保守序列的0.5124 mol。如图1所示, 有41个岛的G+C含量明显高于 (>0.55 mol) 或低于 (<0.45 mol) 基因组0.5089 mol的平均含量, 其中13个是独立的IS元件岛;有54个岛的G+C含量则在0.48 mol到0.53 mol之间。SIs和基因组在某些密码子的使用频率上也存在明显的差异 (图2) , 进一步分析发现这一差异与该岛的G+C含量有关, 即G+C含量与基因组明显不同的岛, 其密码子的使用频率也差异较大。
《图2》
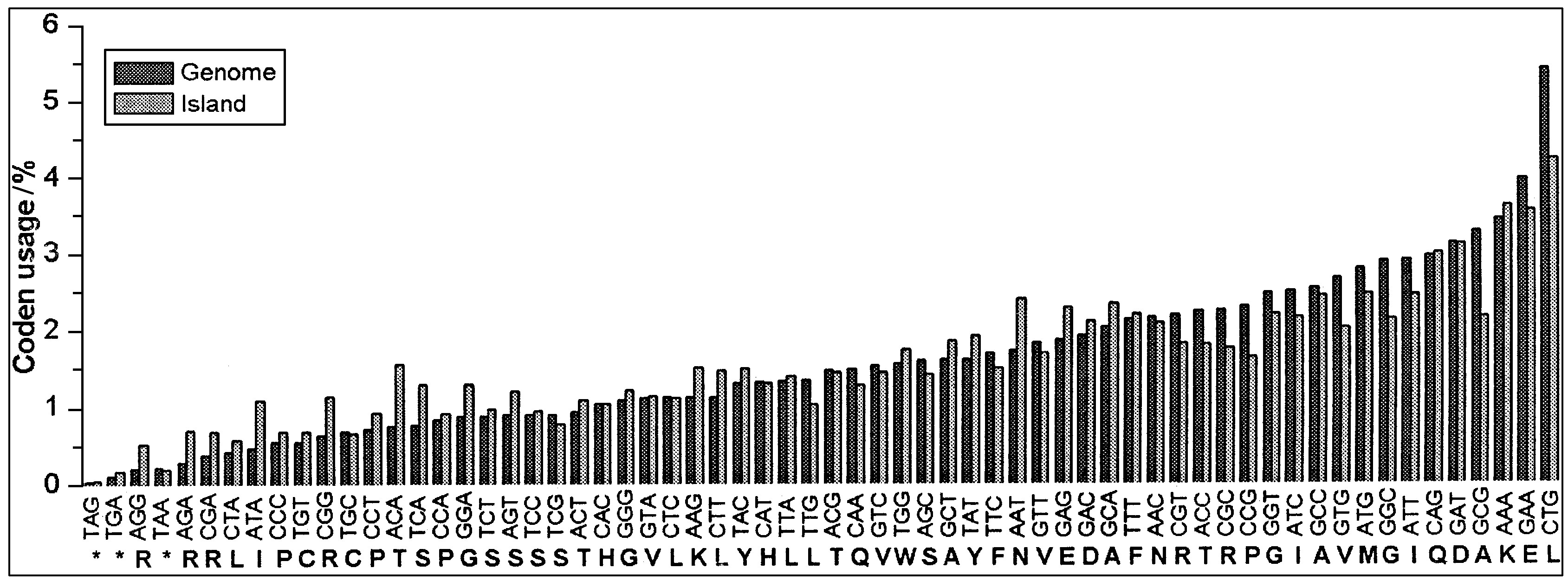
图2 Sf301保守序列与岛上基因的密码子使用频率的分析 (密码子按照在保守序列上的使用频率排列) Fig.2 Codon usage analysis of Sf301 chromosomal genes which on “backbone sequences" (genome) and on SIs (island) , respectively. Codons are put in the order of frequency in the genes on the conserved backbone.
2.3.1 已经报道的PAIs 按照我们的计算方法, 此前在痢疾杆菌中已经鉴定的两个PAIs——SHI-2岛
2.3.2 各SIs的结构分析 按照其ORF的组成, SIs可以分为三组, 第一组有44个, 它们完全或几乎完全由IS元件组成, 称为“IS岛”。该组岛全长81 488 bp, 占SIs的14%;第二组称作“噬菌体相关岛 (phage-related islands) ”, 共31个, 全长近160 Kb, 它们或者完全是噬菌体的残余序列, 或者部分携带有噬菌体相关的ORFs;其余56个岛组成了第三组, 它们均编码其它已知或未知功能的ORFs。
除了有24个SIs的侧翼序列既不是某种氨基酸的tRNA也不是重复序列外, 其余所有107个SIs均与IS元件有关:它们的一侧或者双侧连接有或相同或不同类型的IS元件。所以按照结构特征, SIs可以分为两大类:与IS元件相关的岛和与IS元件无关的岛 。
2.3.3 几个可能的PAIs的预测 侵袭质粒抗原ipa (invasion plasmid) 是痢疾杆菌主要的表面抗原。染色体上有7个ipaH基因的同源序列, 其中2个由于读框内IS元件的插入成为假基因, 另外5个位于我们已经鉴别的岛上。按照在基因组上的排列位置, 我们分别命名为ipaH-岛1-5。
ipaH-岛1由SI 21-26组成, 全长约38 Kb, 插入在Gly-tRNA基因位点;ipaH-岛2由SI 48-51组成, 全长约25 Kb, 两侧均为IS1插入元件, 该岛上有4个连续的ORF与沙门氏菌铁转运系统的sitABCD操纵子相同
SI 1和2组成了一个长约23 Kb, 具有典型毒力岛结构的区域——插入在asp-tRNA位点, 另一侧为IS629插入元件。主要携带有沙门氏菌sci操纵子及一些功能未知的噬菌体ORF的同源序列。
SI 4-10:全长约21 Kb, 虽然序列上有很大的变异, 但仍然保留了痢疾杆菌溶原性噬菌体SfⅡ的主要基因——编码细菌萜醇葡糖基转移酶的bgt和编码葡糖基转移酶Ⅱ的gtrⅡ, 它们是Ⅱ型抗原表达所必须的
SI 83:全长8 178 bp, 两侧均没有特征性结构, 其G+C含量仅为32.49%。该岛主要携带有与福氏痢疾杆菌型特异性O-抗原合成相关的rfb基因簇。
SI 116:全长8 115 bp, 其3'末端紧邻一个IS 1插入元件, G+C含量为38.25%, 主要编码与福氏痢疾杆菌菌体抗原核心多糖生物合成相关的rfa (waa) 基因簇。
《3 讨论》
3 讨论
通过GC偏斜分析以及与大肠杆菌K-12 MG1655的相似性, 确定了Sf301染色体复制原点oriC位点, 它毗邻基因gidB, gidA, mioC 和 asnC, 该区域内包含一段细菌复制特征性的AT-丰富序列。同样的分析显示, Sf301染色体的复制终止点 (terC) 位于染色体的1.60 Mb 附近, 在环型染色体上, 该区域位于复制原点的对面, 从而将其分成两个大致相等的复制子, 表明了染色体的对称结构。虽然在大肠埃希氏属中可以允许有低水平的染色体非对称性
自1950以来, 志贺菌一直被作为一个属来研究, 并被分成了四种:痢疾志贺菌 (S.dysenteriae) 、福氏志贺菌 (S.flexneri) 、宋内氏志贺菌 (S.sonnei) 及鲍氏志贺菌 (S.boydii)
痢疾杆菌的致病过程主要包括细菌到达结肠粘膜, 侵入粘膜上皮细胞并在细胞内繁殖, 同时扩散到相邻细胞, 引起程序性细胞死亡, 最终造成肠粘膜水肿破坏并脱落。侵袭性大质粒是痢疾杆菌最主要的毒力因子, 与其致病性密切相关
脂多糖 (lipopolysaccharide, LPS) 是革兰氏阴性菌的主要表面成分, 由类脂A、核心多糖和O-特异性多糖链即O-抗原三部分连接而成, 其中O-抗原的特异性是革兰氏阴性菌血清型分型的基础。已知染色体上与his操纵子连锁的rfb基因簇与福氏痢疾杆菌型特异性O-抗原合成相关
痢疾杆菌主要的致病特点是侵袭结肠粘膜上皮细胞, 虽然侵袭相关基因都定位于毒力大质粒上, 但其毒力基因的完全表达却受染色体上多个基因的调控











 京公网安备 11010502051620号
京公网安备 11010502051620号




