

天然葡激酶 (Staphylokinase, Sak) 是金黄色葡萄球菌溶原性噬菌体合成的一种蛋白水解酶, 由136个氨基酸组成。Sak本身并不是酶, 它在人血浆中与纤溶酶原 (plasminogen, plg) 形成1∶1复合物, 该复合物被血块表面痕量的纤溶酶 (plasmin, plm) 激活为Sak·plm, Sak·plm是高效的纤溶酶原激活剂, 激活游离的plg形成plm, 催化血栓主要基质纤维蛋白降解, 从而溶解血栓。由于Sak激活plg具有纤维蛋白专一性, 而且对陈旧性血栓和富含血小板血栓的溶解作用比其他溶栓药物更强, 因此Sak是一种很有希望的溶栓剂
在研究过程中发现, 葡激酶易形成二聚体, 甚至多聚体。聚合体的形成很可能导致Sak抗原性的增强, 分子量 的增加亦不利于口服制剂的研制, 而且国家生物制品规程规定, 聚合体的比例一般不能超过蛋白质总量的10%。因此, 研究防止二聚体形成的新型葡激酶分子显得十分必要。
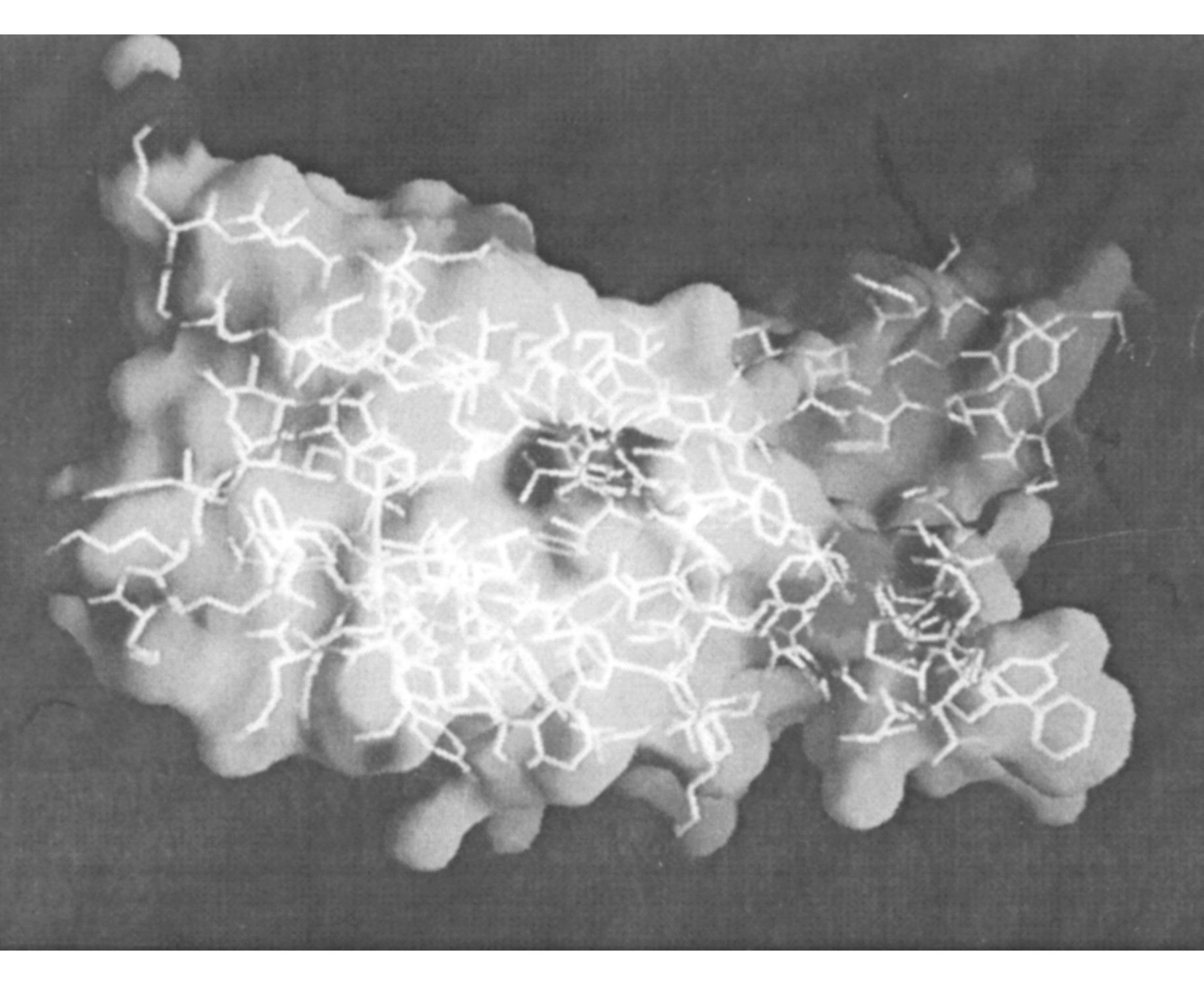
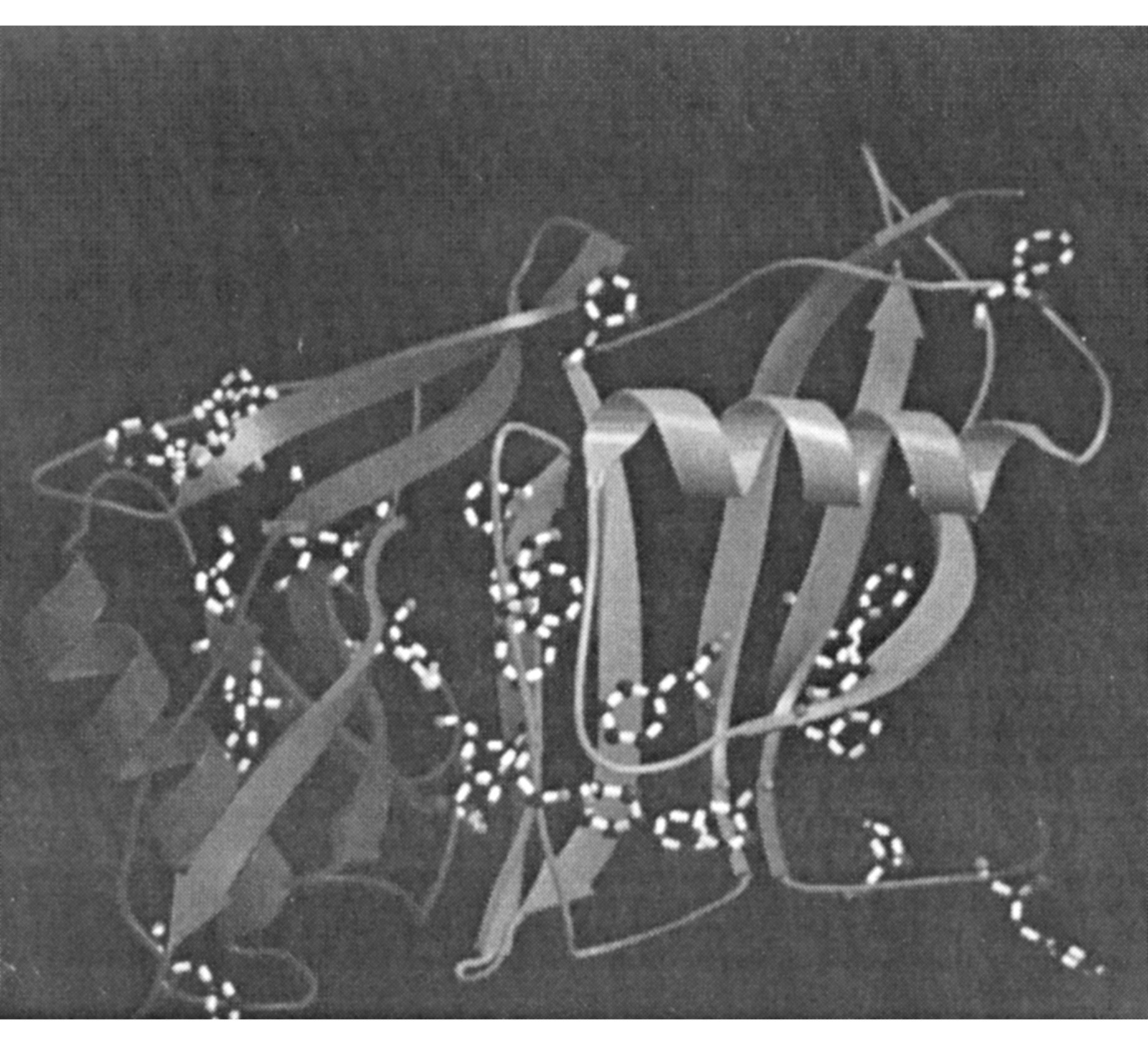
与清华大学饶子和教授合作, 利用X射线晶体衍射技术, 已经建立了Sak精确的三维结构模型。Sak是一椭球状分子, 从第21个氨基酸残基起, 分别由5条、2条β折叠股构成的β折叠片包裹在一个由12个残基构成的α螺旋上, 而N端20个氨基酸伸向球体外, 非常柔韧, 其功能难以从晶体结构推测。Sak呈明显的亲疏水性的不对称性, 且活性区主要在亲水一侧。
在此结构基础上, 通过计算机分子对接方法, 预测了二聚体可能的结合方式, 建立了二聚体的三维结构模型, 由此设计了突变体RGD-Sak, 并实现了在大肠杆菌中的高效表达及高度纯化。突变体RGD-Sak与野生型Sak相比, 纤溶活性虽然有所下降, 但是形成聚合体的能力明显降低, 这提示建立的二聚体结构模型基本合理, 可在RGD-Sak的基础上进行进一步的改造, 以期得到不产生聚合作用而活性保持的新型葡激酶分子。
《1 材料与方法》
1 材料与方法
1.1材料
1.1.1 合作与开发 与清华大学饶子和教授合作, 测定了本室研制的r-Sak的晶体结构。分子对接软件是美国Rockefeller大学I.A.Vakser开发的GRAMM V1.03
1.1.2 菌种与质粒 大肠杆菌JM109、质粒pUC19为本室保存。大肠杆菌JF1125、原核表达载体pLY-4由中科院生物化学研究所刘新垣教授惠赠。质粒pST-Sak为本室构建。
1.1.3 试剂与仪器 Sak半成品由本室提供 (970923) , 原纯度达98%以上, -70℃保存。ExpandTM High Fidelity PCR System购自BM公司, 核酸工具酶购自BRL公司, Qiagen核酸纯化柱购自基因公司, 丙烯酰胺、尿素购自Sigma公司, S-Sepharose FF、ImageMasterR VDS购自Pharmacia公司。5L发酵罐、蛋白质层析系统分别为美国NBS、Waters公司产品。其余试剂均为国产分析纯。
1.1.4 寡聚核苷酸 由美国Johns Hopkins大学DNA合成组制备。
1.2方法
1.2.1 r-Sak半成品的SDS-PAGE电泳鉴定 还原性、非还原性SDS-PAGE, 参照分子克隆实验指南进行。翻转酪蛋白凝胶板法活性测定, 参见文献
1.2.2 葡激酶二聚体的分子模拟 为了确定Sak二聚体的结合区, 以GRAMM V1.03软件进行分子对接, 选择一分子Sak为受体, 另一分子Sak为配体, 以Sak受体搜索与配体Sak的结合区。按笔者推荐的整体高分辨率对接参数进行对接 (表1) , 搜索了10个复合物的结构。
表1整体高分辨率对接参数
Table 1 The parameters for high-resolution
generic docking
Matching mode (generic/helix) mmode=generic
Grid step eta=1.7
Repulsion (attraction is always-1) ro=30
Attraction double range (fraction of single range) fr=0
Potential range type (atom radius, grid step) crang=atom radius
Projection (blackwhite, gray) ccti=gray
Represention (all, hydrophobic) crep=all
Number of matches to output maxm=10
Angle for rotations, deg (10, 12, 15, 18, 20, 30, 0-no rot)
ai=10
1.2.3 RGD-Sak基因的克隆及原核表达质粒的构建
设计克隆引物:
上游引物5′...CGC GAA TTC ATC TCA AGT TCA TTC GAC...3′
下游引物5′...CGC GGA TCC TTA TTT CTT TTC...3′
突变引物5′...TAA ATC TGG GAC GAC GTC ACC ACG TTC TGT TAT AGG...3′ 引入PstI位点
质粒pST-Sak为模板, 以上游引物、突变引物进行第一轮扩增, 351 bp扩增片段经琼脂糖凝胶电泳回收、纯化后, 与下游引物再次以质粒pST-Sak为模板进行第二轮扩增。纯化后, 所得408 bp片段为模板, 以上游引物、下游引物进行第三轮扩增, 产物经Klenow酶补平, EcoRI、BamHI酶解后与pUC19重组, 酶解筛选阳性克隆, 核苷酸序列分析验证是否发生预计位置的突变, 由基康生物技术公司用ABI377测序仪完成序列分析。然后用EcoRI、BamHI将RGD-Sak基因切出, 连入表达载体pLY-4的相应位点。
1.2.4 工程菌的诱导表达 将pLY-4-RGD-Sak转化大肠杆菌JF1125, 筛选高表达菌株, 然后以5L发酵罐进行低密度发酵, 温度诱导后, 离心收集菌体, PBS洗涤后, -70℃保存待用。
1.2.5 包涵体的分离、溶解与复性 湿菌用PB缓冲液悬浮, 以高压匀浆泵压榨, 离心后, SDS-PAGE分析目的蛋白的存在状态。
上述沉淀经洗涤, 离心后, 以0.1 mol/L PB、8 mol/L尿素、0.5% β-巯基乙醇溶解, 室温作用至澄清透明。超离后弃沉淀, 取上清稀释复性。
1.2.6 S-Sepharose FF柱层析 以10倍柱体积PB缓冲液平衡色谱柱, 稀释复性后溶液直接上柱, Waters色谱仪控制流速和检测蛋白峰。上样结束后, 以PB缓冲液洗至基线, 0~1 mol/L NaCl梯度洗脱, 收集洗脱组分, SDS-PAGE分析目的蛋白分布, 并测定蛋白浓度。
1.2.7 纯度鉴定及分子量测定 样品进行15%SDS-PAGE, 考马斯亮兰R-250染色后, Pharmacia Imagemaster VDS扫描测定纯度、分子量。
1.2.8 生物学活性测定 分别以纤维蛋白凝胶板溶圈法、酪蛋白凝胶板溶圈法、发色底物法测定。
1.2.9 Sak·纤溶酶复合物和RGD-Sak·纤溶酶复合物的Km和Kcat值测定 参照文献
1.2.10 聚合能力检测实验 野生型Sak为对照, 以生理盐水溶解, 取30 mg/mL、3 mg/mL高、低两种蛋白质浓度, 室温静置。每24 h取样, 电泳鉴定。
《2 结果与讨论》
2 结果与讨论
2.1 r-Sak半成品蛋白质组成分析、活性测定
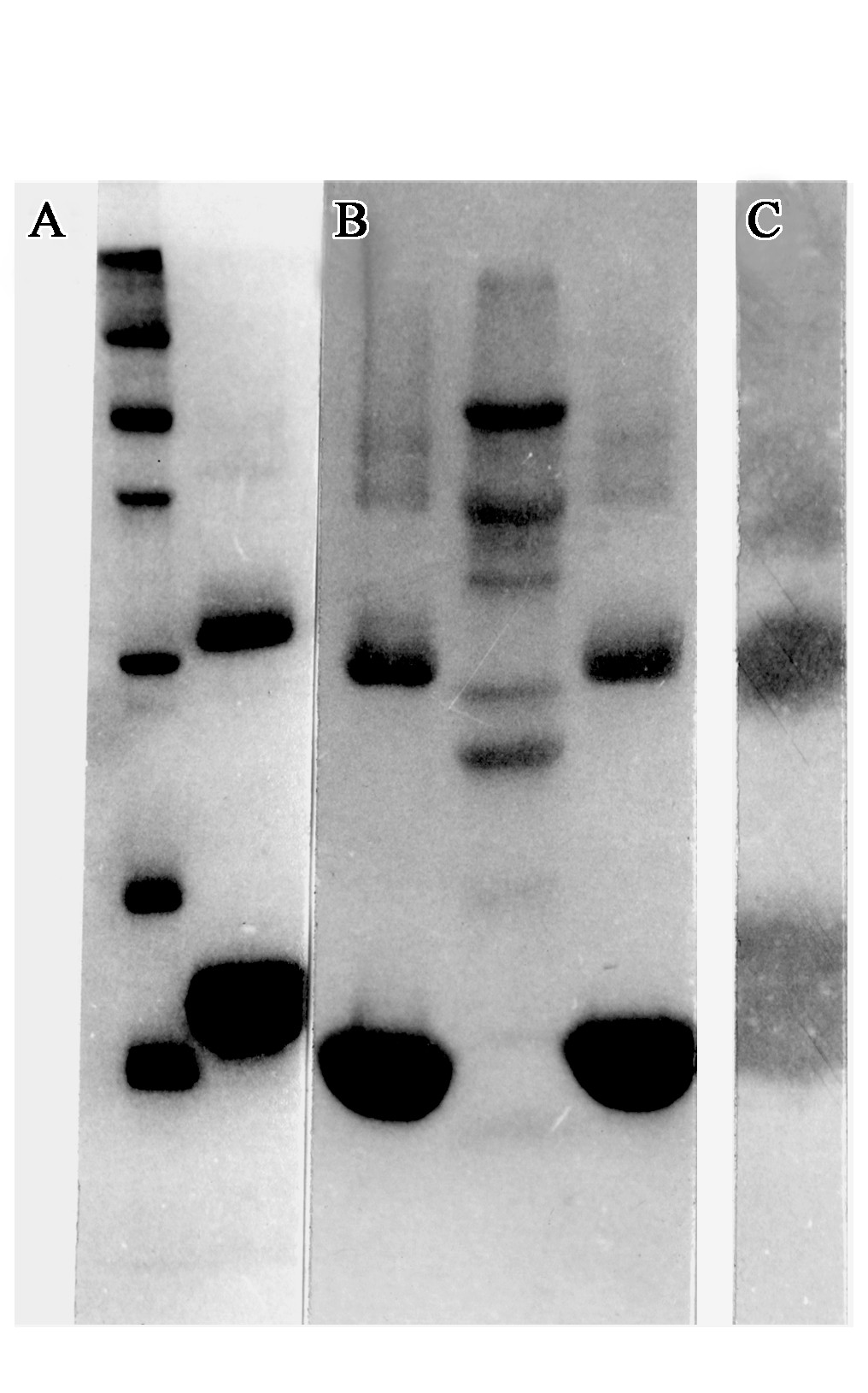
分别以还原性、非还原性SDS-PAGE电泳分析半成品蛋白质组成。电泳结束后, 以考马斯亮兰染色, 在相对分子量约15.5 kD、31 kD、46 kD、62 kD处出现浓集的条带 (图1) 。翻转酪蛋白凝胶板法测定活性, 在各条带相应位置均有清亮的透明溶解带。表明r-Sak半成品储存过程中有形成抗SDS聚合体的倾向, 且聚合物非常稳定, 并保持一定活性。
2.2 Sak二聚体的分子模拟及突变体设计
静电势及疏水性分析表明, Sak单体呈明显的亲疏水性的不对称性 (图2) 。Silence等通过随机突变的研究表明, 决定活性的氨基酸主要位于亲水一侧
《图1》
《图2》

图1还原性、非还原性SDS-PAGE分析r-Sak及翻转酪蛋白凝胶板法测定活性
Fig.1 The reducing, non-reducing SDS- PAGE and zymography analysis of r-Sak (A) reducing SDS-PAGE:1—Marker (14.4 kD、20.1 kD、31 kD、43 kD、66 kD、97.4 kD) ;2—r-Sak (B) non-reducing SDS-PAGE:1, 3 r-Sak;2—Marker (14 kD、17 kD、24 kD、29 kD、36 kD、45 kD、66 kD) (C) Zymography:1—r-Sak
蛋白质相互作用的界面通常有6~13 nm2 (600~1300 2) , 每个蛋白质提供10~30个接触残基, 但是, 界面中存在所谓“热点区”, 仅3~5个氨基酸就提供了80%左右的结合能, 改变这些残基, 复合物的结合能力将显著下降
2.3 RGD-Sak基因克隆及原核表达质粒构建
第一轮PCR扩增后, 琼脂糖凝胶电泳分析PCR产物, 扩增片段约351 bp。第二轮扩增得约408 bp的片段, 酶切证实已引入PstⅠ位点。第三轮扩增后的产物与pUC-19重组, 转化大肠杆菌, 酶解筛选阳性克隆, 核苷酸序列分析证实已发生设计的突变。
目的基因与pLY-4重组, 转化大肠杆菌JF1125, 提取质粒, 以EcoRⅠ、BamHⅠ酶解, 得约408 bp条带;BamHⅠ、PstⅠ酶切, 得约340 bp条带, 证实获得阳性克隆。
2.4 RGD-Sak在大肠杆菌中的表达与鉴定
质粒pLY-4-RGD-Sak转化大肠杆菌JF1125, 经温度诱导表达, SDS-PAGE分析表达产物。电泳结束后, 一半进行考马斯亮兰染色, 可见诱导后细菌裂解液在分子量约15.5 kD处有一浓集的条带, 经扫描, 重组蛋白约占全菌总蛋白的50%;另一半洗去SDS后, 贴在酪蛋白凝胶板上, 37℃孵育数小时后, 相当于15.5 kD处有一明显的透亮区, 即此部位的酪蛋白已溶解, 表明RGD-Sak具有纤溶活性。菌体经压榨离心后, 15.5 kD条带主要位于沉淀中, 而上清中几乎未见此条带, 说明表达产物以包涵体形式存在。
2.5 RGD-Sak的分离纯化
10 L发酵液得湿菌80 g。取20 g压榨破菌, 离心, 得包涵体5 g。经包涵体洗涤、溶解、稀释复性后, 大部分杂蛋白已被洗去或在复性过程中沉淀, 经S-Sepharose FF柱一步纯化至均质。
2.6激活纤溶酶原试验
RGD-Sak.plasmin激活纤溶酶原的反应符合米氏方程 (表2) 。
表2RGD-Sak.plasmin、Sak.plasmin激活纤溶酶原的酶促动力学常数比较
Table 2 Kinetic analysis of activation of plasminogen by Sak moieties
《表2》
| Km/mol·L-1 | Kcat/s-1 | Kcat/Km | |
Sak.plasmin | 8.91 | 0.96 | 0.11 |
RGD-Sak.plasmin | 12.40 | 0.81 | 0.07 |
由表2可见, RGD-Sak的活性有所下降, 可能突变仍对活性区造成了影响。另一方面, Sak在同一系统中高效表达时, 为可溶性蛋白质, 而RGD-Sak却形成了包涵体, 复性时折叠不够完全, 可能也是活性下降的原因。因此, 仍需进一步优化分子结构, 摸索复性条件。
2.7聚合能力检测试验
在两种蛋白质浓度下, RGD-Sak均显示了较Sak弱的聚合能力。Sak的前10个氨基酸易被降解, 因此可见部分降解条带。采用生理盐水建立聚合的快速检测模型, 是为了更接近生理条件。
曾试图分离纯化二聚体, 进行酶动力学和X射线晶体衍射的研究, 以建立结合区的精确模型。但是, 后来发现单体、聚合体随盐浓度变化呈动态平衡关系, 聚合体在去盐过程中又部分解离, 因此无法得到适合于结晶的高纯度无盐样品。本文得到的RGD-Sak的聚合能力明显降低, 而且可能具备抑制血小板聚集的功能, 有一定的临床应用前景, 同时也表明二聚体X射线晶体结构得到解析之前, 利用计算机模拟进行分子设计, 是一种值得尝试的手段。











 京公网安备 11010502051620号
京公网安备 11010502051620号




