

《1 前言》
1 前言
当今,全球水资源短缺和水环境污染问题日益严重,海水淡化处理技术在保障国计民生方面意义越来越重大。反渗透法凭借其能耗低,处理效率高,出水水质好的优点,已经占世界海水脱盐产水市场份额的一半以上[1] 。反渗透膜材料作为该技术的核心,经过半个多世纪的发展,已经由早期的醋酸纤维素膜发展到当前主流的聚酰胺复合反渗透膜[2] 。由于聚酰胺复合膜是在超滤支撑膜(如聚砜膜)上通过界面聚合的方法形成一层厚度在200 nm 左右致密的聚酰胺分离层,膜厚远小于相转化制备的醋酸纤维素膜,因此膜的渗透阻力明显下降,节约了运行成本,从而促进了反渗透技术在海水淡化处理领域的大规模应用。
聚酰胺膜虽然具有水通量大,截盐率高的优点,但在实际运用中也存在一些问题,如海水中因含有大量微生物,在待处理过程中需要添加活性氯抑制水中微生物的繁殖,而聚酰胺材料易被活性氯破坏,虽然目前的工艺是在反渗透处理的前端使用活性碳等吸附剂去除余氯[3] ,但未除尽的余氯仍会缓慢地破坏聚酰胺膜,使膜的分离性能逐渐下降,缩短了膜的使用寿命。因此增强反渗透膜的耐氯性对于延长膜使用寿命,简化预处理工艺具有非常重要的意义。
《2 聚酰胺膜氯化降解理论》
2 聚酰胺膜氯化降解理论
活性氯破坏聚酰胺膜的机理仍不十分明确,但随着对一些先进表征手段被应用于该研究,逐渐对该过程形成了一些共识:a.活性氯在水中存在的形式根据pH的变化而不同(见表1),酸性条件下,主要以Cl2分子形式存在,在中性或偏碱性条件下,以 HClO的形式存在,在过碱性条件下,以ClO-离子形式存在;b.当Cl2为主要形式时,发生的氯化反应是 Cl2对聚酰胺多元胺侧的苯环的不可逆直接氯取代; c.当HClO为主要形式时,发生的氯化反应是HClO 先对酰胺键N—H的可逆氯化,随后再发生不可逆 Orton 重排,使酰胺键上的 Cl转移到多元胺侧的苯环上(见图1);d.ClO-为主要形式时,基本不发生氯化反应[4,5] 。氯化后,聚酰胺分子间的氢键(由酰胺键形成)遭到破坏,导致聚酰胺层由规整的结晶态向无定型态转变,分子链的变形将增大聚合物膜的自由体积,从而使大尺寸的盐离子可以透过膜[6] ;同时氯化后的酰胺键将逐渐水解断裂,导致膜的交联度下降[5,7] ;膜被氯化后与支撑低膜发生分离,使膜内产生空隙,这些都将导致膜分离性能的恶化[8] 。
《表1》
表1 25 ℃活性氯组分分数与pH的关系[9]
Table 1 Effect of pH on the composition of free chlorine at 25 ℃[9]
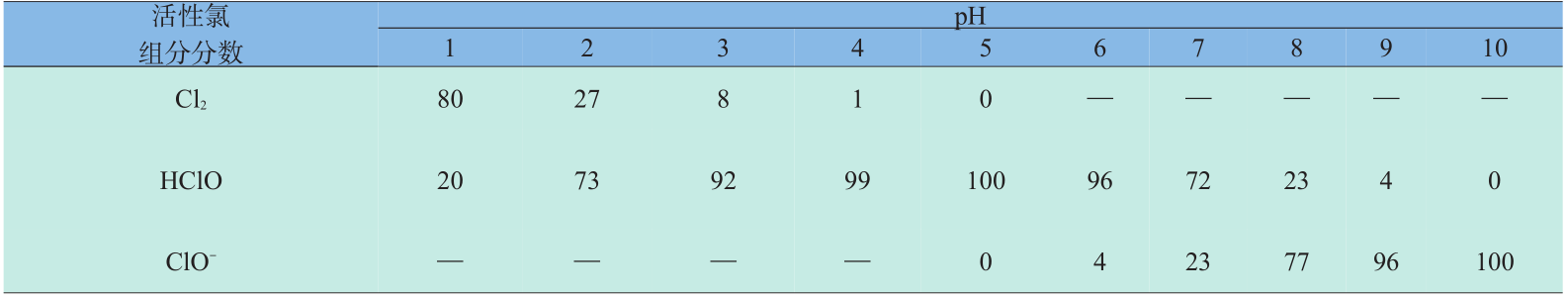
《图1》
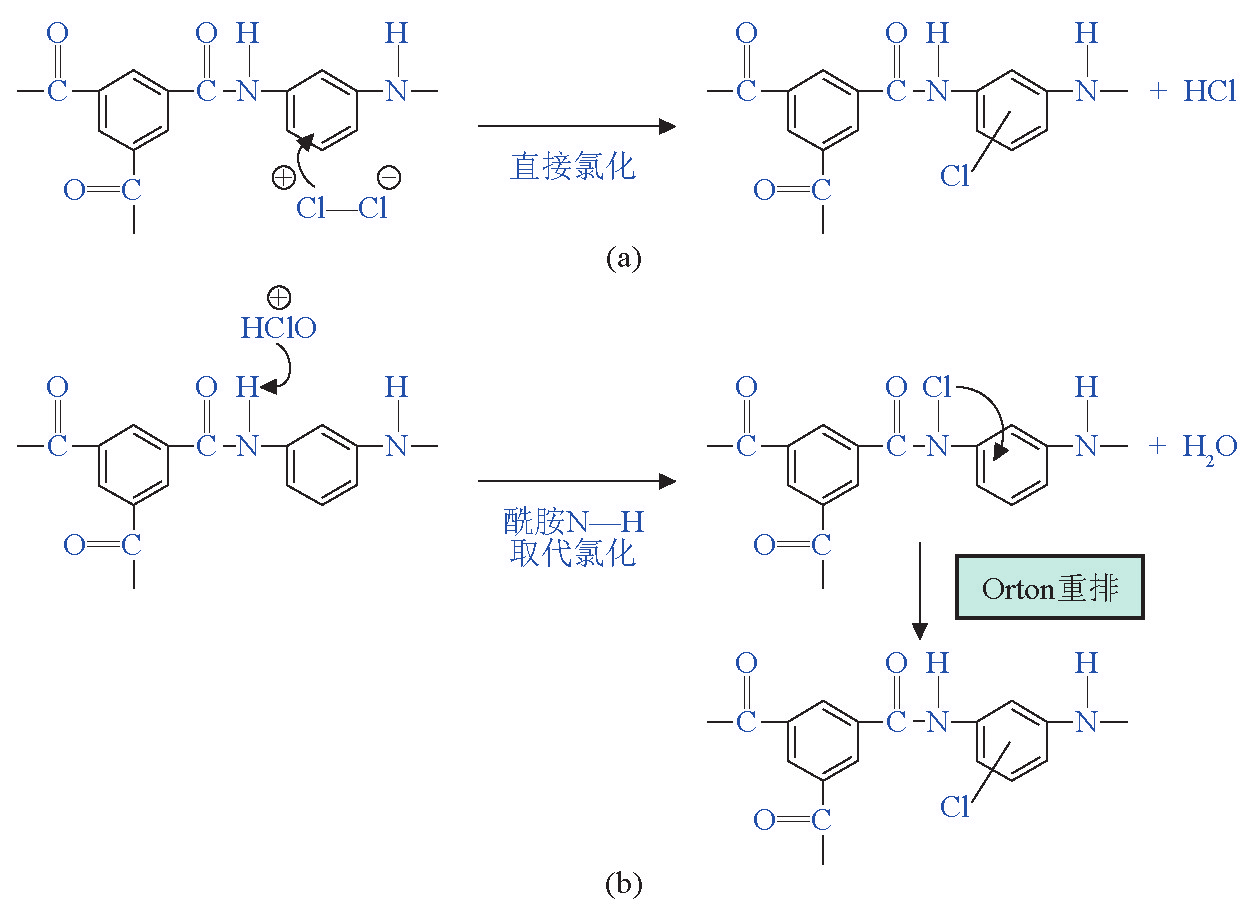
图1 聚酰胺膜氯化机理图
Fig. 1 Chlorination mechanism of polyamide membrane
《3 耐氯反渗透膜的研究方法》
3 耐氯反渗透膜的研究方法
几十年来,为了解决反渗透膜耐氯性不佳的问题研究者们进行了广泛的研究,改进方法主要包括以下三种。
《3.1 表面涂覆保护层》
3.1 表面涂覆保护层
在聚酰胺表面涂覆一层亲水聚合物材料(如 PVA[2] 、PEG[10] )不仅能有效增强膜的耐氯性还能提高膜的抗污染性能,但表面涂覆层增加了膜的渗透阻力,而且涂覆层在长期的运行过程中可能发生脱落失效的现象。
《3.2 表面修饰》
3.2 表面修饰
利用聚酰胺膜表面具有酰氯水解后产生的羧酸基团,对聚酰胺膜表面进行化学改性修饰,如引入含有与活性氯优先发生反应的基团的聚合物作为牺牲层[11,12] ,或者接枝聚合物保护层防止活性氯与聚酰胺上活性位点的直接接触[13,14] ,与简单的涂覆法相比该方法能提高保护层对聚酰胺层的吸附能力,延长有效保护时间。
《3.3 开发本体耐氯的聚合物膜材料》
3.3 开发本体耐氯的聚合物膜材料
与前两种方法相比,开发新的耐氯反渗透膜材料能从根本上增强反渗透膜的耐氯性。但是要开发耐氯性良好且具有反渗透分离性能的膜材料难度较大,目前主要分为三类:即磺化聚砜材料、叔酰胺聚合物和修饰化仲酰胺聚合物材料。本文总结了近年来新型本体抗氯材料的主要研究进展(见表2)。
《表2》
表2 本体耐氯材料及其耐氯能力
Table 2 Chlorine-tolerant materials and their durability under chlorination treatment
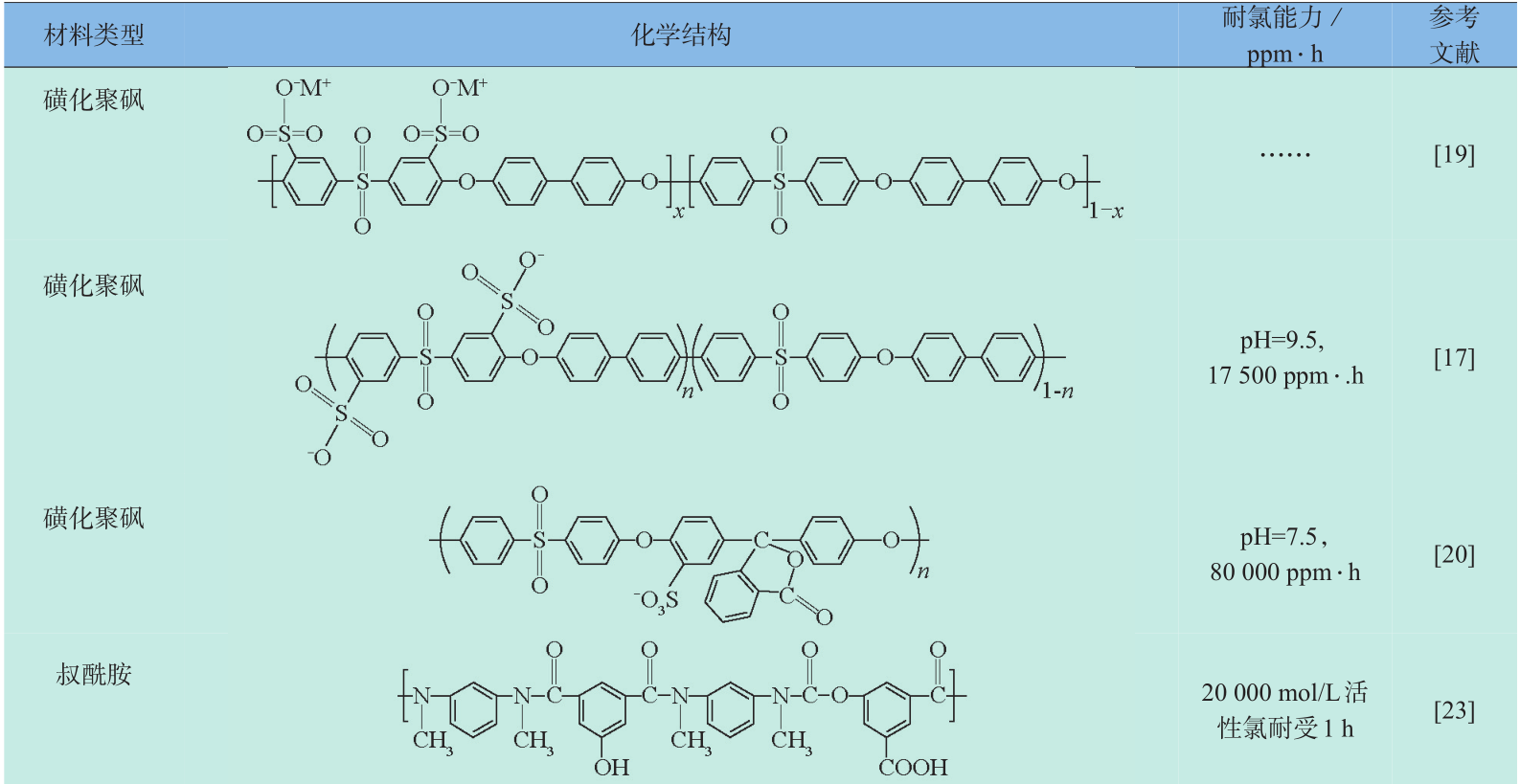
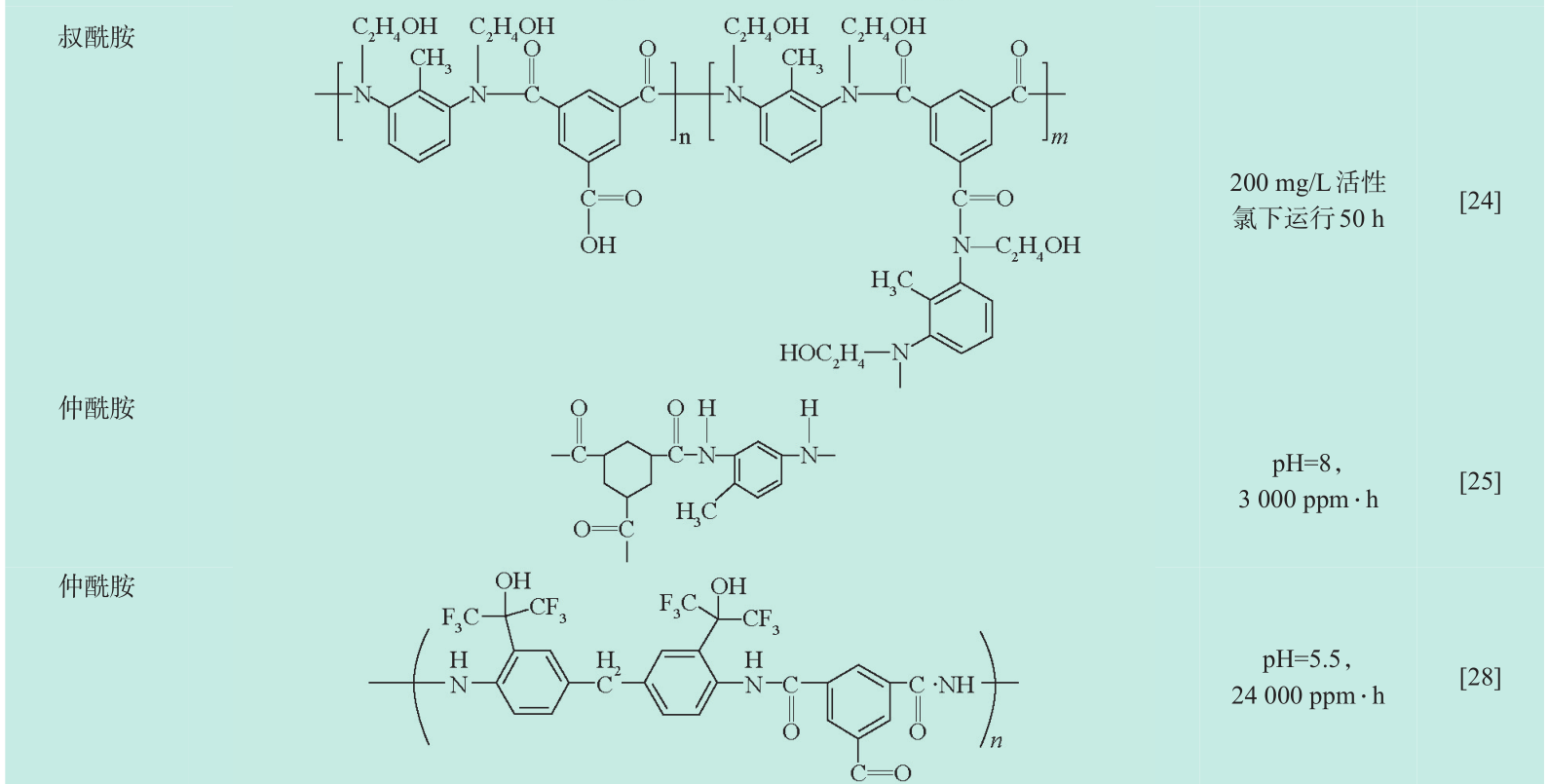
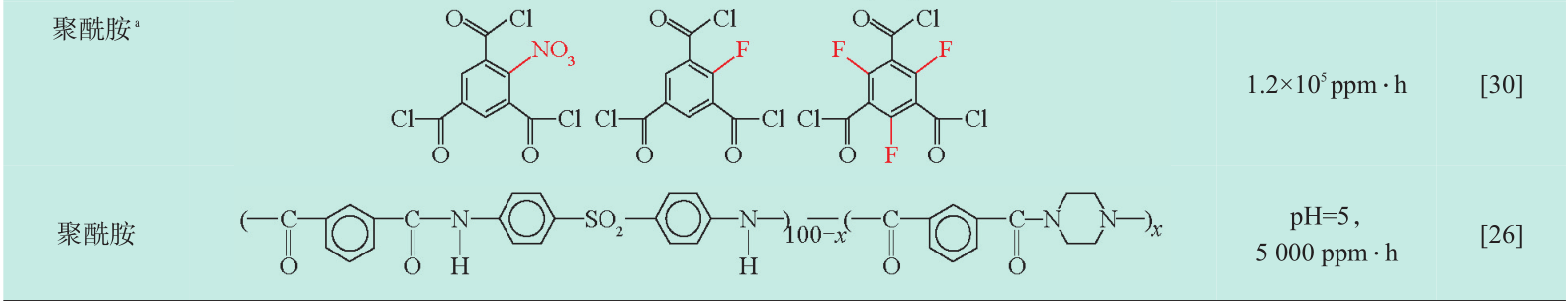
注:耐氯能力(ppm ⋅ h):活性氯浓度(ppm)× 处理时间(h);a 为使用的改性的酰氯单元(红色为修饰基团);1 ppm=10-6
3.3.1 磺化聚砜类材料
聚砜材料原本是制备成微孔膜作为聚酰胺反渗透复合的底膜,但由于聚砜分子链中不含活性氯攻击位点(如聚酰胺膜中的酰胺键),耐氯性能优异,是目前为数不多的商品化非聚酰胺类耐氯反渗透材料。1973年,Quentin[15] 首次发现致密的磺化聚砜膜同样具有盐水分离特性,随着磺化程度的增加,膜的水渗透性增加但截留率下降,因此合理的磺化处理是制备该类反渗透材料的关键。Cadotte[16] 使用磺化剂处理了含有两类苯环结构(砜基苯环与双酚 A 苯环)的聚砜,发现磺化反应优先发生在双酚 A 苯环上,当磺化剂进一步增加时,砜基苯才会发生部分磺化反应,说明通过调控磺化剂可以实现可控的磺化反应。但由于该材料早期的制备方法是先聚合(得到聚砜)再磺化,导致磺化反应的不均一,强酸性的磺化剂(如硫酸或氯磺酸)导致一些不利的副反应发生(如主分子链断裂、过度的交联),针对这些问题Park[17] 采用了先制备磺化单体,再聚合的策略,通过调节聚合中使用的磺化/非磺化单体的比例,实现了对聚砜材料磺化度的精确调节。随后Paul[18] 同样采用先磺化单体再聚合的方法制备了高度磺化的聚砜膜材料,同时针对过度磺化将损失截盐性能的缺点,使用MY721型环氧树脂对制备的磺化聚砜膜进行交联,交联后磺化聚砜膜的截盐率从73.4 %上升到97.2 %。Stevens[19] 为了改善该类膜厚度大,渗透阻力高的缺点,在BPS-20型磺化聚砜的铸膜液中加入NH3NO3,发现NH3NO3可以有效促进相转化过程中溶剂交换速率,获得厚度为纳米级的致密的分离层,分离试验结果表明膜的渗透阻力明显下降。Zhang[20] 在超滤聚砜底膜上制备一层厚度 400 nm左右的酚酞型聚芳醚砜,随后在分离层表面涂覆PVA亲水层以提高膜的抗污染和耐氯性,制备得到了截留率达 96.8 %,可在 2 000 ppm 活性氯下稳定运行36 h的反渗透膜。
磺化聚砜对盐离子的截留机理尚无统一认识,但磺酸基团的引入明显改善了原聚砜膜疏水的缺点,有助于提高膜通量,同时有实验表明处理体系中引入二价钙离子会降低该类膜对一价盐离子的截留[21] ,鉴于钙离子具有遮屏Donnan渗析膜上电荷的效应,说明Donnan效应可能是该类膜截留盐离子的机理之一。磺化聚砜膜通常采用相转化法制备,导致膜的厚度大,渗透阻力高,因此需在高压下运行,与主流的聚酰胺膜相比能耗较高,尚难以大规模应用于水处理工业中。
3.3.2 叔酰胺材料
叔酰胺聚合物膜由于酰胺键 N 上没有易被活性氯取代的H,耐氯性较传统仲酰胺材料大为提高,Konagaya[22] 分别使用脂肪族仲胺、环形脂肪族仲胺及邻位取代的对苯二胺与间苯二甲酰氯进行聚合,发现仲胺与酰氯生成的叔酰胺比对苯二胺形成仲酰胺聚合物具有更强的耐氯性。刘立芬[23] 使用N, N-二甲基间苯二胺与 5-氯甲酰氧基-异肽酰氯制备了反渗透膜,当活性氯浓度从 4×103 mol/L 增加到 2×l04 mol/L时,该膜的脱盐率和通量几乎没有变化,而且与未浸泡的膜相比,其截留率和通量甚至还有部分上升,这可能是由于活性氯将该类膜内残余的仲胺单体氧化,从而使膜产生了“紧致效应”,提高了膜的截留率,而仲胺被氯氧化之后可能生成醌类物,这类物质能与水分子缔合,增加了膜的亲水性,提高了膜的水通量;Zhang[24] 使用双-2,6-N, N-(2-羟乙基)甲苯二胺与间苯三甲酰氯进行界面聚合聚合在聚砜底膜上制备了叔酰胺聚合膜,不仅氨基能与酰氯能反应形成酰胺键,羟基也能与酰氯形成酯键,N上的酯键能减低酰胺键上的电子云密度,从而减低了酰胺 N 与活性氯的取代反应活性,制备的纳滤膜能在8 000 ppm ⋅ h活性氯处理后保持 80 %的 MgSO4截留率。叔酰胺聚合膜通过引入取代基团的方法阻断酰胺N被活性氯进攻的路径,但同时也破坏了聚酰胺类反渗透膜内的分子氢键,聚合物分子的致密度下降,使得这类膜的截留性能变化比较明显,取代基团体积越大,分离膜分离性能也越趋近纳滤范围。
3.3.3 修饰化仲酰胺聚合物
鉴于酰胺的N—H结构在形成分子间氢键中具有重要作用,许多研究选择在不取代酰胺键的N— H结构的前提下对聚酰胺材料进行化学修饰。由于聚酰胺膜的氯化主要发生多元胺一侧,故对多元胺单体的基团修饰是提高聚酰胺膜抗氯性的重要方法之一,通过引入大尺寸的官能团,增大立体位阻阻碍氯原子从酰胺N上通过Orton重排反应转移到苯环上;或者通过引入强吸电子基团,降低苯环上电子云密度,以降低苯环与活性氯发生的亲电取代,从而提高膜对活性氯的耐受性。Yu[25] 使用4-甲基间苯二胺与1,3,5-三甲酰氯环己烷进行界面聚合,制备得到了耐氯极限能达3 000 ppm· h,并具有反渗透性能的聚酰胺膜(1 500 ppmNaCl 进料液浓度,1.5 MPa 操作压力下截留率达到 97.5 %,通量 52 L/(m2 · h));Konagaya[26] 使用 4,4′-二胺基二苯砜和哌嗪的无规共聚物与间苯二甲酰氯聚合制备得到一种线形嵌段共聚物,再使用相转化的方式制备得反渗透膜,膜的截留率也能达到99 %以上,耐氯性能也明显强于聚间苯二甲酰-间苯二胺膜,能在 50 ppm的活性氯溶液中能稳定运行100 h 以上,但是水通量很小(2.71 L/(m2 ⋅ h)),这可能是由于相转化法制备的膜厚度较大,渗透阻力大造成的;随后他又采用3,3′-二氨基二苯砜与对苯二甲酰氯及3,5-二胺基苯甲酸的共聚物制备得到了一种线性结构的聚砜酰胺分离膜,其耐氯性比常规的聚酰胺反渗透也有明显提高,但水通量仍然较低[27] ;La [28] 使用3,3′-双(1-羟基-1-三氟甲基-2,2,2-三氟乙基)-4,4′-甲基二苯胺为多元胺单体与均苯三甲酰氯界面聚合制备得到了对NaCl截留能力大于90 % 的反渗透膜,三氟甲基的引入不仅增大了重排的立体位阻,而且该基团强烈的吸电子能力能有效地降低苯环的电子云密度,降低苯环发生亲电取代的活性,X射线光电子能谱分析(XPS)分析显示,氯化处理后该聚合的含氯量仅为 0.6 %,而传统的间苯二胺与均苯三甲酰氯聚酰胺反渗透膜聚合物在相同氯处理条件下含氯量达 5.6 %,且由于该聚合物强吸电子的三氟甲基影响了与之相邻的羟基,使其变得易于电离,因此该分离膜的性能明显受到进料液的pH条件的影响,pH升高时羟基发生电离,使得膜的亲水性增强,水通量增大,同时Donnan排斥效应增强,导致膜对盐离子的截留性能也提高;Han[29] 使用了三聚氰胺作为多元胺反应单元,将传统的苯环单元用三嗪环单元取代,有效地增强了膜的抗氯性,但截留率较差,只能达到纳滤的分离效果。
此外,对酰氯单元的一些结构调控仍能明显改变膜对活性氯的耐受性,Murphy[30] 在酰氯的苯环上应引入了氟、硝酸根、氯等吸电子基团,利用这些修饰化的多元酰氯单元(如单氟聚苯三甲酰全氟聚苯三甲酰氯,全氯均苯三甲酰氯或硝酸基均苯三甲酰氯与常见的多元芳香胺通过界面聚合的方法制备了高耐氯性的聚酰胺反渗透膜,能在24 h内在1.2× 105 ppm· h的活性氯条件下稳定运行。
《4 结语》
4 结语
近年来耐氯膜材料的开发主要集中在新型聚酰胺合成单体的开发和对现有聚酰胺膜的改性(如界面修饰和表面涂覆)上,但由于对聚酰胺膜氯化降解机理的研究仍不十分明确,根据现有理论的指导尚难以彻底解决聚酰胺膜的耐氯性问题,而非聚酰胺类反渗透膜在通量或截留率上难以超越聚酰胺膜,尚没有很好的商业化产品问世。为了进一步提升拓展反渗透海水脱盐技术的应用范围,除了需要继续深化对聚酰胺膜氯化机理的研究外,还需在此基础上不断推进相关耐氯聚酰胺反渗透膜的合成与制备研究,此外也需要加快非聚酰胺类反渗透膜材料的开发,为下一代反渗透膜的开发奠定基础。











 京公网安备 11010502051620号
京公网安备 11010502051620号




