
2002, Volume 4, Issue 10
Strategic Study of CAE >> 2002, Volume 4, Issue 10
Complete Genome Sequence of Shigella flexneri 2a 301 Strain and Analysis of “Shigella-islands”
1. State Key Laboratory for Molecular Virology and Genetic Engineering, Beijing 100052, China
2. Laboratory of Molecular Virology, Fudan University, Shanghai 200032, China
3. Institute of Epidemiology and Microbiology , Chinese Academy of Preventive Medicine , Beijing 102206, China
4. Peking University Health Science Center, Beijing 100083, China
5. National Center of human Genome research, Beijing 100176, China
6. Huabei Pharmaceutical Co. , Ltd. Shijiazhuang 050000, China
7. Microbial Genome Center, Ministry of Public Health , Beijing 100052, China
Next Previous
Abstract
Shigella flexneri serotype 2a are the most prevalent species and serotype that cause .bacillary dysentery or shigellosis in man. This paper presents the complete genome sequence of a Shigella flexneri 2a strain which isolated from the Beijing outbreak, and the primary analysis of “ Shigella-genomic islands (SIs),” that means Shigella flexneri 2a 301 strain-specific genome fragments. The whole genome is composed of a 4,607,203 bp chromosome and a 221,618 bp virulence plasmid, designated pCP301. The chromosome shares a conserved ‘backbone’ sequence about 4.03 Mb with those of a benign laboratory strain E. coli K12 (MG1655) which is essentially collinear. Sf301 has 572 Kb specific-sequence which form into 320 Sis with sizes greater than 50 bp and encoding in total 519 Shigella-specific Open Reading Frames (ORFs). Among these Sis, there are 131 islands with sizes greater than 1 Kb with repeated sequences of transposable elements, transposons or tRNAs flanking on one or both sides. The average G + C content of the Sis is 48.25% , significantly lower than that of the conserved backbone. Frequency of codons such as ACA, AAT, GCG, CTG, etc. , on Sis are quite distinct from that on backbone sequences. All above observations together suggest that many of the Sis are foreign origin. Among them, the authors identified 7 putative Sis with typical structure of pathogenicity islands (PAI) and 2 Sis harbor some ORFs related to biosynthesis of lipopolysaccharide (LPS) have implications in virulence, in addition to the previously identified PAIs, SHE and SHI-2. The other Sis are mostly a mosaic of genes of known function and ORFs encoding polypeptides sharing none or low homology with known proteins from one or more bacterial species. All of these could be subjected to investigations towards novel preventive and treatment strategies against shigellosis.
Figures
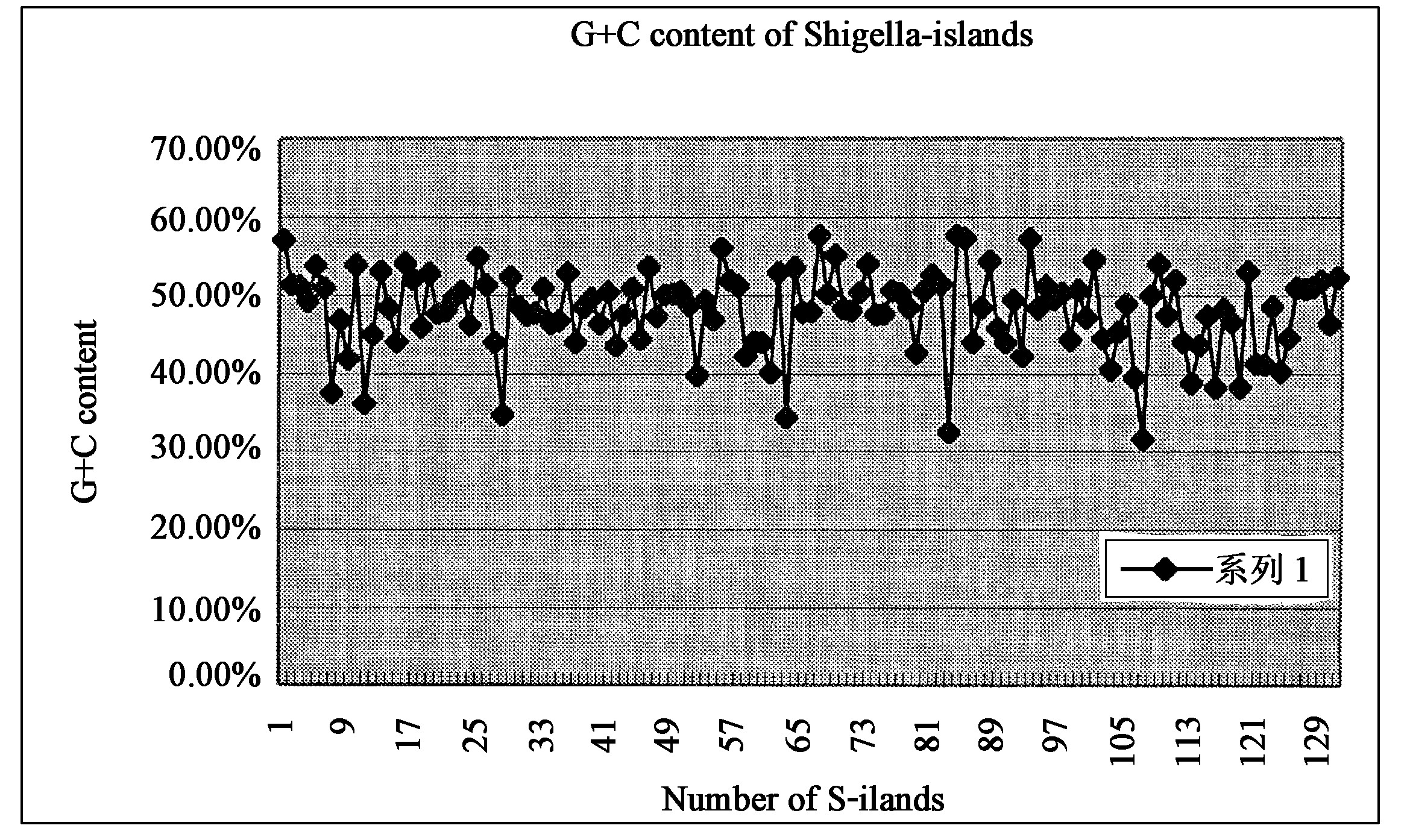
图1
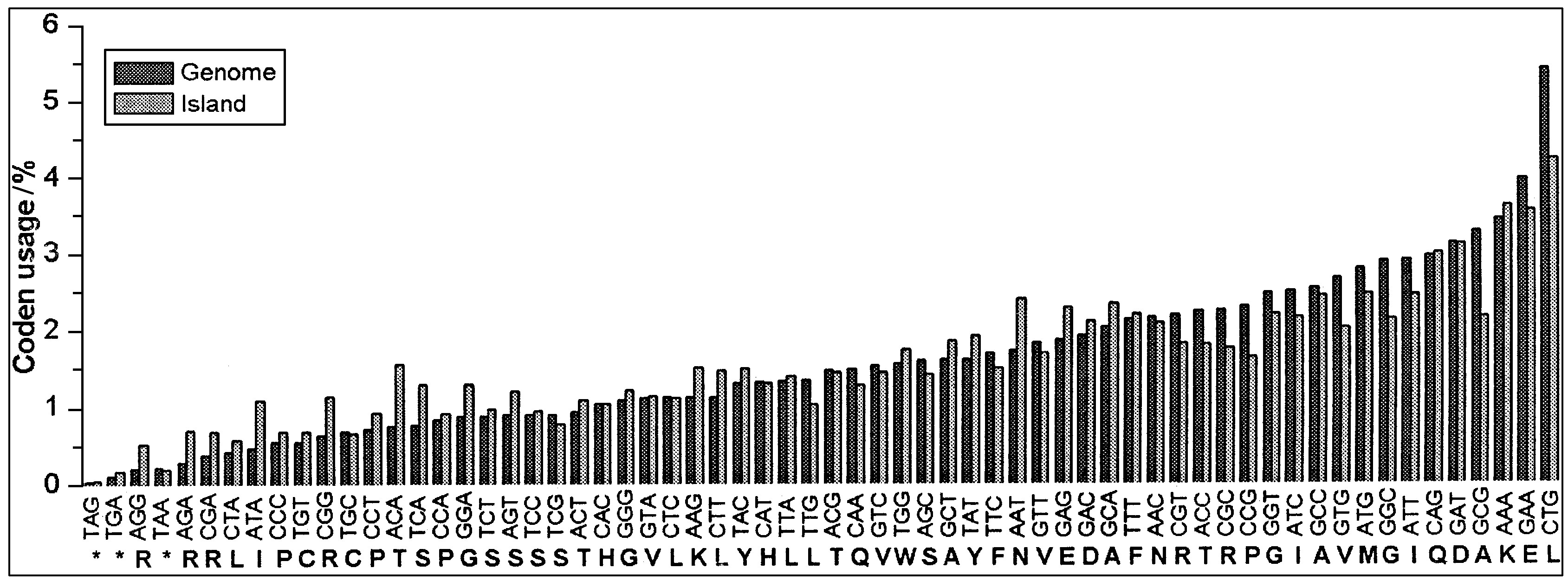
图2
References
[ 1 ] SansonettiPJ.Microbesandmicrobialtoxins:paradigmsformicrobial mucosalinteractionsⅢ.Shigellosis:fromsymptomstomolecularpathogenesis[J].AmJPhysiolGastrointestLivPhysiol, 2001, 280, G319~G323
[ 2 ] MeiY , LiuH , XuJ.CloningandApplicationofgenusspecificDNAprobesforshigella[J ].ChineseJEpidemiol, 1989, 10:167~170
[ 3 ] LeeCA .Pathogenicityislandsandtheevolutionofbacterialpathogens[J].InfectAgentsDis.1996Jan, 5 (1) :1~7
[ 4 ] KarlinS .Detectinganomalousgeneclustersandpathogenicityislandsindiversebacterialgenomes[J].TrendsMicrobiol2001Jul, 9 (7) :335~343
[ 5 ] AusubelFM , etal.颜子颖等译.精编分子生物学实验指南[M ].北京:科学出版社, 1998, 39 link1
[ 6 ] EwingB , HillierL , WendlMC , etal.Base callingofautomatedsequencertracesusingphred.I .Accuracyassessment[J].GenomeRes, 1998, (8) :175~185
[ 7 ] GordonD , AbajianC , GreenP .Consed:A graphicaltoolforsequencefinishing[J].GenomeRes, 1998, (8) :195~202
[ 8 ] SalzbergSL , DelcherAL , KasifS , etal.MicrobialgeneidentificationusinginterpolatedMarkovmodels[J].NucleicAcidsRes, 1998, 26, 544~548
[ 9 ] TatusovRL , GalperinMY , NataleDA , etal.TheCOGdatabase:atoolfor genome scaleanalysisofproteinfunctionsandevolution[J].NucleicAcidsRes, 2000, 28, 33~36
[10] LoweTM , EddySR .tRNAscanSE :aprogramforimproveddetectionoftransferRNA genesin genomicsequence[J].NucleicAcidsRes, 1997, 25, 955~964
[11] PlattnerFR ., PlunkettGIII, BlochCA , etal.ThecompletegenomesequenceofEscherichiacoliK -12[J].Science, 1997, 277, 1453~1474
[12] MossJE , CardozoTJ , ZychlinskyA , etal.TheselC associatedSHI 2 pathogenicityislandofshigellaflexneri[J].MolMicrobiol, 1999, 33, 74~83
[13] RajakumarK , SasakawaC , AdlerB .Useofanovelapproach, termedislandprobing, identifiestheShigellaflexnerishepathogenicityislandwhichencodesahomologoftheimmunoglobulinAprotease likefamilyofproteins[J].InfectImmun, 1997, 65, 4606~14
[14] JanakiramanA , SlauchJM .The putativeirontransportsystemSitABCDencodedonSPI1isrequiredforfullvirulenceofsalmonellatyphimurium[J].MolMicrobiol2000Mar;35 (5) :1146~55
[15] PernaES , PlunkettGⅢ, BurlandV , etal.GenomicsequenceofenterohaemorrhagicEscherichiacoli0157:H7[J].Nature, 2001, 409, 529~533
[16] MavrisM , ManningPA , MoronaR .MechanismofbacteriophageSfII mediatedserotypeconversioninshigellaflexneri[J].MolMicrobiol, 1997, 26, 939~950
[17] TetsuyaH , KozoM , MakotoO , etal.CompletegenomesequenceofenterohemorrhagiceschelichiacoliO157:H7and genomiccomparisonwithalaboratorystrainK 12[J].DNAResearch2001, 8, 11~22
[18] BergthorssonU , OchmanH .DistributionofchromosomelengthvariationinnaturalisolatesofEscherichiacoli[J].MolBiolEvol1998Jan, 15 (1) :6~16
[19] LiuGR ., RahnA , LiuWQ , etal.TheevolvinggenomeofSalmonellaentericaserovarPullorum[J].JBacteriol2002May, 184 (10) :2626~33
[20] HaleTL .Geneticbasisofvirulenceinshigellaspecies[J].MicrobiolRev, 1991, 55, 206~224
[21] PupoGM , LanR , ReevesPR .Multipleindependentoforiginsofshigellaclonesofescherichiacoliandconvergentevolutionofmanyoftheircharacteristics[J].ProcNatlAcadSciUSA , 2000, 97, 10567~10572
[22] SansonettiPJ , KopeckoDJ, FormalSB , etal.Involvementofaplasmidintheinvasiveabilityofshigellaflexneri[J].InfectImmun1982Mar, 35 (3) :852~60
[23] SasakawaC , KamataK , SakaiT , etal.Virulence associatedgeneticregionscomprising31kilobasesofthe230 kilobaseplasmidinShigellaflexneri2a[J].JBacteriol, 1988, 170:2480~2484
[24] BuysseJM , HartmanAB , StrockbineN , etalGeneticpolymorphismoftheipaHmulticopyantigengeneinshigellaspps.andenteroinvasiveescherichiacoli[J].MicrobPathog, 1995, 19:335~349
[25] RajaumarK , JostBH , SasakawaC , etal.NucleotidesequenceoftherhamnosebiosyntheticoperonofShigellaflexneri2aandroleoflipopolysaccharideinvirulence[J].JBacteriol1994Apr, 176 (8) :2362~2373
[26] OkadaN , SasakawaC , TobeT , etal.Virulence associatedchromosomallociofshigella flexneriidentifiedbyrandomTn5insertionmutagenesis[J].MolMicrobiol1991Jan, 5 (1) :187~95
[27] FormalSB , GemskiP , Jr, etal.GenetictransferofshigellaflexneriantigenstoescherichiacoliK 12[J].Infect.Immun, 1970, 1, 279~287
[28] SansonettiPJ , HaleTL , DamminGJ , etal.AlterationsinthepathogenicityofescherichiacoliK 12aftertransferofplasmidandchromosomalgenesfromshigellaflexneri[J].InfectImmun1983Mar, 39 (3) :1392~402











 京公网安备 11010502051620号
京公网安备 11010502051620号




