
1. 引言
电子器件材料的计算研究在引入新功能材料中发挥着关键性的作用,以满足对器件尺度的要求。传统场效应晶体管(FET)器件由Si半导体通道、硅化物金属电极源极/漏极和带有多晶硅金属栅的SiO2绝缘栅极介电层组成,用于对通道进行现场控制。在过去十年间,Si基器件材料被迅速取代:高介电常量(高 κ)氧化物代替了SiO2,而金属栅代替了多晶硅栅[1]。在这个快速进程中,材料设计发挥着关键性的作用,指引人们从许多候选材料中选择极具潜力的高 κ 电介质和金属栅材料[2–5]。最近,美国政府启动了材料基因组计划(MGI),旨在推动先进工程系统应用新材料的发展与商业化[6]。MGI以在新功能材料开发中引入理性材料设计为目标,试图缩短材料开发周期(大大缩短了传统上20年的周期),而非遵循传统依靠实证试错的方法。值得注意的是,笔者的研究工作(关于另一个主题)已经证实从概念设计开始到商业产品开发用时在八年以内,从而证实了MGI的理念[7–9]。从这个角度看,FET器件扩展的下一步是引入高迁移率通道材料来代替Si通道,其属于MGI问题范畴。具有预测性的量子模拟通过关注材料设计所选定的最有前途的材料系统,为缩短开发周期提供关键性的指导。在本文中,笔者对高迁移率通道与高 κ 栅氧化物间界面所形成的俘获态起源进行了讨论,从而将FET器件面临的挑战控制在MGI框架范围内。
高 κ 氧化物与III-V通道之间的低界面质量仍然是制造超高速半导体器件的主要障碍之一。尽管改进微观过程、钝化、自清洁和表征还需要做大量工作[10–13],对界面态的物理起源、高 κ 氧化物/GaAs界面中的费米能级钉扎的有限理解阻碍了在提高界面质量方面的突破性进展。这种阻碍主要归因于复杂的界面结合形式;根据实验生长条件,界面结合形式不同。在高κ氧化物/GaAs界面中,Ga(As)可能诱发各种结构无序,如Ga-(As-)氧化物、Ga—Ga(As—As)二聚物键合、Ga—(As—)悬挂键等,这些结构无序可能会降低界面的绝缘性质。在自清洁效应中,(NH4)2S和NH4OH能还原As-氧化物,而且最近通过特定的原子层沉积(ALD)过程也能还原As-氧化物[14]。因此,剩余缺陷对隙间态分布的影响变得非常关键[15]。实验证明,界面态密度(Dit)被分为三个部分,即P1、P2和P3,如图1所示。Dit分布的起源仍是一个活跃的研究领域。例如,长期以来对Ga3+是否会导致隙间态仍存在争议[14–16]。除Ga3+外,当不同数量的O原子与Ga成键时,3+与1+之间的Ga部分电荷态将会出现。这些部分电荷态可与Ga3+和1+共存,也有可能成为隙间态的来源之一。如果没有充分理解不同Ga电荷态对界面态分布的影响,隙间态分布机制则有可能不完整。此外,氧化态的存在可能标志着界面结合无序,这会导致更多缺陷的产生,如悬挂键。
从理论上来说,依据块体GaAs的经验性紧束缚分 析,如图1(b)所示,Ga— (As—)悬挂键生成导(价)边缘 态(VB:P1和CB:P3)。As—As (Ga—Ga)二聚物和Ga (As)错位形成中间隙间态,如图1(b)中的P2所示[17]。 然而,由于未考虑界面结合,这种模型没有充分洞察GaAs/氧化物界面隙间态的起源。事实上,在高κ/GaAs 界面中,HfO2等高 κ 氧化物的离子键合没有固定的原子配位和固定的键角,这导致界面Ga和As产生了不同电荷态。这些电荷态与不同类型的不饱和Ga (As)键相符,从而产生隙间态。
《图1》
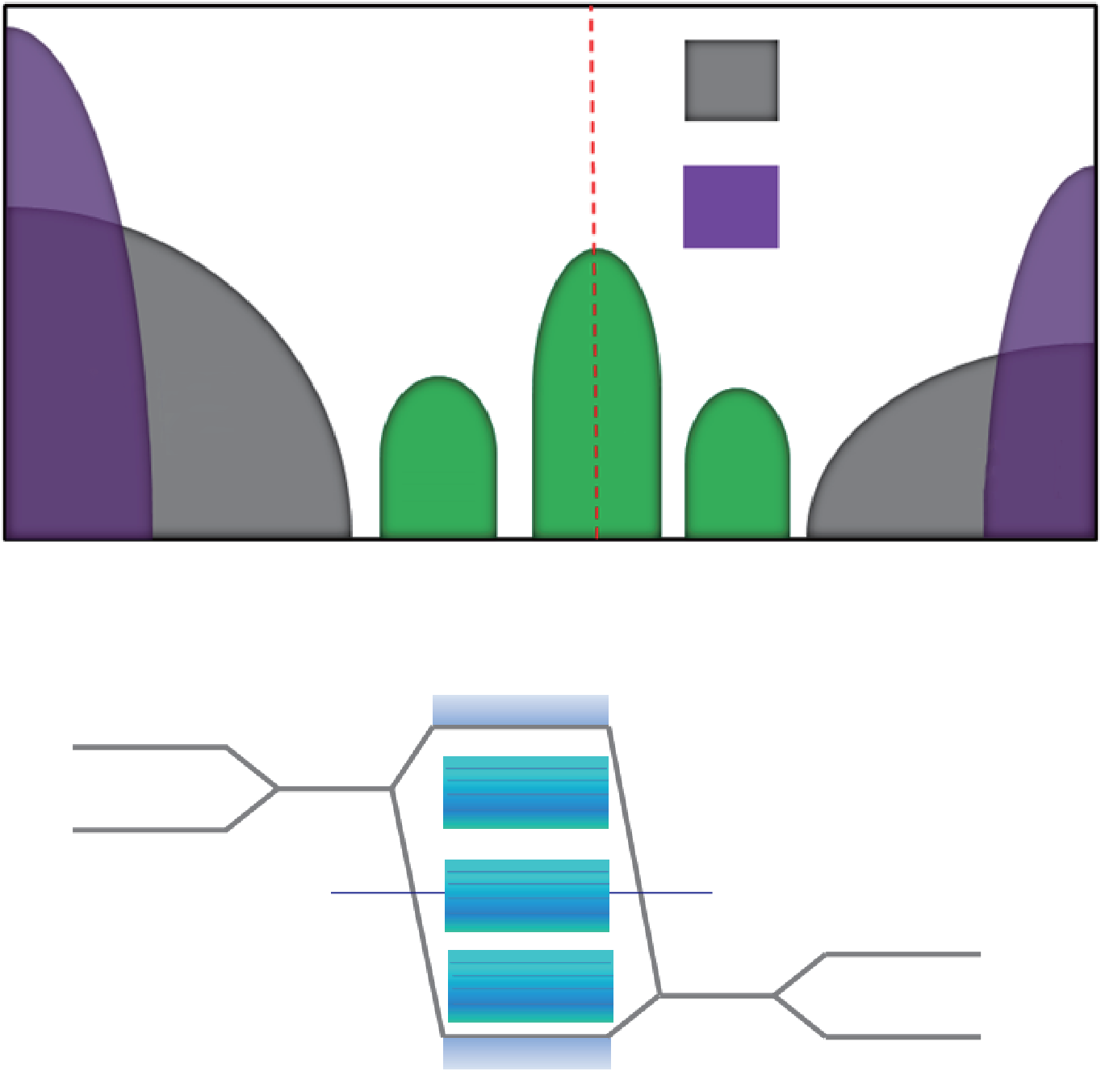
图 1. (a) 界面态密度 (Dit) 被分为三个部分,即 P1、P2 和 P3;(b) GaAs缺陷能级的紧束缚分析。VB 为价带;CB 为导带;VBM 为价带顶; CBM 为导带底。
从本质上来说,很难得到没有任何结构不完整性的高κ/GaAs电子突变界面,原因如下:①GaAs(001)表面呈极性;②Ga呈三价,而As呈五价。每个Ga—As中有0.75个电子源于Ga,有1.25个电子源于As,这导致电子计数规则、Ga(As)共价键角和方向性的严格需求难以被满足[18]。这种情况意味着部分带电的Ga(As)键合会产生界面隙间态。最近,Robertson等[19]提出了由1×1单胞的 GaAs和 HfO2表面构建的(001)方向GaAs/HfO2界面,并得出结论,即以O替换到下面GaAs层中的As位上,有可能获得一个绝缘界面。然而,这个界面模型包含了一个由HfO2(5.07Å) 和 GaAs(5.65Å) 间大晶格失配引起的大界面平面应变(>10%),这使得模型界面问题重重。
在未重构的以Ga(As)终止的GaAs表面,原子形成了一个方阵列。每个表面Ga(As)有两个指向表面外的部分占据的悬挂键。根据Ga(As)—O共价键的严格要求,Hf和O都不能直接钝化Ga—(As—)悬挂键,除非在界面上施加大平面应变,而这在实践上是不现实的。替代方法之一是系统性地模拟界面氧气键合产生的效应[20]。这种键合的检测和与高 κ 电介质处理过程或表面处理相应的氧化态已经被实验充分证实[21]。
2. 结果与讨论
以标记为O10的界面模型为例,如图2所示,其中每个氧层有10个O原子,每个Ga(As)层有4个Ga(As)原子。因此,为了形成HfO2绝缘面,HfO2表面需要5个O原子。在GaAs表面有8个Ga—悬挂键,需要3个O原子进行钝化。因此,界面O10以界面处的2个多余O原子终止。移走这2个多余的O原子就可生成中性界面。尽管整个界面具有电中性,3个O原子却不一定均匀地向每个Ga键分配1.25个电子,因此这种情况必然会形成隙间态,同时捕获费米能级。此外,从界面处移走2个界面Ga原子和5个O原子后,可以建立另一个中性界面,在此之后GaAs表面和HfO2将呈绝缘状态。然而,这第二个界面是不稳定的,因为其Ga—O键合度低于带有全部O终止的界面。
《图2》
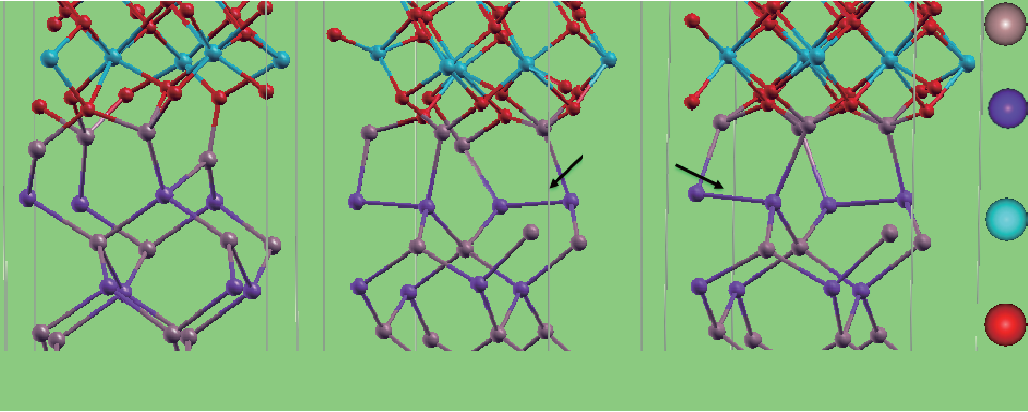
图 2. (100) 界面O8、O9 和O10 的侧视图。Ga、As、Hf 和O 原子分别用灰色、紫色、浅蓝色和红色球表示[20]。
影响界面稳定性的一个重要参数是外部氧的化学势,其控制界面氧化并在根本上影响界面稳定性。例如,对于最先进的ALD生长技术而言,界面稳定性在很大程度上依赖于氧化剂(O3、O2和H2O[22,23])的浓度。为探索外部氧化学势对界面氧浓度的影响,依次移走界面的1~7个O原子[20],界面形成能是氧化学势的函数,而氧化学势的范围约束在O2和HfO2之间(图3)。这种探索表明带有9个界面O原子的界面(在图2中标记为O9/Ga4)在很大的一个氧化学势范围(~49.0%)内具有最低的形成能。此外,笔者发现有8个界面O原子的中性带电界面只在整个生长条件21.6%的范围内才能保持稳定。界面带有2个Ga原子和5个O原子的另一个中性带电界面在整体氧化学势范围内显然是不稳定的。相反,~88.4%的生长条件范围内产生了一个非中性带电界面O9。这一观察结果解释了原子和电子突变界面不可能自发出现的原因。
《图3》
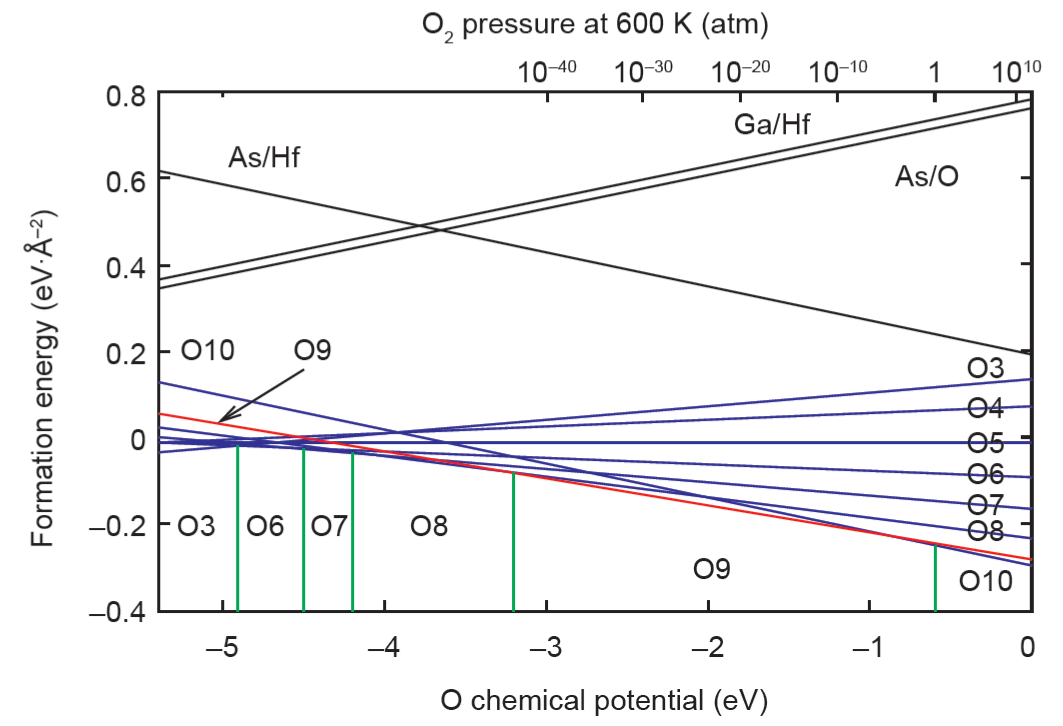
图 3. 作为氧化学势的函数的不同界面结构的界面形成能。Ga—O 键合:根据界面 O 原子数量,标记为 O3~O10。As—Hf 键合:As/Hf。As—O 键合:As/O。Ga—Hf 键合:Ga/Hf [20]。1 atm = 101.325 kPa。
根据富氧条件,氧化物的形成未被严格控制,因此O8、O9和O10界面是最有现实意义的,如图2所示。O9界面是在很大的一个氧化学势范围内最稳定的模型,GaAs第二(As)原子层和第三(Ga)原子层之间的一个Ga—As被打断,并从Ga—O界面键经过优化自发地形成一个Ga—悬挂键和两个相距分别为两层和一层的As—As二聚物。O10界面中也发现了类似效应。有趣的是,由于界面存在中性电荷,O8界面具有最少的“错误键合”(即As—As和Ga—悬挂键)。为进一步检查Ga和As的电荷态,进行了Bader电荷[24]计算,如图4(a)和(b)所示。对于O10模型界面而言,界面Ga电荷态接近3+。由于耗尽了O10界面的氧,在O9和O8模型中,Ga的更多的部分电荷态开始出现,如图4(a)所示。对于As电荷态而言,发现了O9和O10界面的As—As二聚物电荷态,如图4(b) 所示。
《图4》


图 4. 晶体内部及O8、O9和O10界面(a)Ga和(b)As的电荷分布
图5为利用Heyd-Scuseria-Ernzerhof(HSE)混合密度泛函计算得到的GaAs块体(灰色区域)及O8/Ga4、O9/Ga4和O10/Ga4界面模型的态密度(DOS)。用远离界面的Ga(As)原子表示块体的DOS。在块体GaAs间隙区域发现了三种导致费米能级钉扎的界面态。为探索隙间态的起源,图5中的插图显示了间隙区域内的局域电荷分布。由于O9/Ga4在很大的一个氧化学势生长条件范围内具有最高的稳定性,所以笔者对界面Ga和As原子的电荷态进行了检查(图4)。笔者将4个界面Ga原子G1、G2、G3和G4分别标记为电荷态1.64e、1.63e、2.23e和1.63e。局域电荷清楚地表明界面态P1、P2和P3分别由Ga(G2)部分氧化、Ga—悬挂键和As—As二聚物产生。Ga(G1、G3和G4)3+对隙间态没有直接贡献。
《图5》

图 5. 块体 GaAs ( 灰色区域 ) 和界面 O8、O9 和 O10 的 DOS。块体的 DOS 被放大了两倍,且标准化了界面 DOS。在块体 GaAs 间隙区域内的界面态用粉红色进行了填充。VBM 和 CBM 分别用 VB 和 CB 边缘表示。 费米能级设定为 0 eV。插图表示间隙区域内的局域电荷分布。O8、O9 和 O10 等值线间距为 4.0 × 10–2 eV·Å–3。
本质上,由于强的电负性,界面氧吸引着相邻Ga原子的电子,因而形成了Ga3+、Ga1+等Ga电荷态和一些界面态。界面Ga下面的As原子向其上部键合原子提供的电子多于向下部键合原子提供的电子。为了补偿As的电荷损失,会形成As—As二聚物。笔者发现,Ga—悬挂键和As—As二聚物的形成极大地依赖于界面Ga的电荷态。具体而言,大电荷损失引起的Ga3+的形成进一步推动了As的大电荷损失,因此As形成了As—As二聚物。实验证明,Ga3+和As—As二聚物的共存使我们难以识别隙间态的真正起因。笔者假设界面Ga电荷态降为1+(Ga2O),其提供的电荷小于3+;由于可以移走界面As、As—As二聚物和Ga—悬挂键,从而有望相应地消除隙间态[25,26]。这个发现为有效界面钝化机制提供了一个光明的线索。
非晶Si(a-Si)的使用似乎是一个有效的钝化策略。如果界面存在a-Si,Si向界面Ga提供电荷,并将Ga部分氧化态转化为1+;同时,As—As二聚物从Si获得足够的电荷来打破二聚物键[27]。
除界面热稳定性和DOS外,注入势垒是半导体器件的另一个临界参数[28],用栅氧化层的VB边缘与半导体VB边缘之间的能带偏移表示。在器件应用中,注入势垒需要大于1.0eV,以防止电子进入氧化层的CB,进而使载流子穿透栅氧化层[29]。
利用参考电位法可以准确预测价带偏移(VBO)[30,31]。在界面模型O9中,VBO等于1.81eV。相比之下,实验数据给出了2.00eV[32]、2.10eV[33]和2.85eV[34]的多元化数值。Robertson和Falabretti在没有考虑所观察到的界面结合的情况下采用电荷中性能级(CNL)法,预测出3.00eV[35]的数值。这些发现明确了HfO2和GaAs之间大有希望的注入势垒。
总而言之,为了发现HfO2/GaAs界面态(即As—As二聚物、Ga部分氧化态和Ga—悬挂键诱导的隙间态)的起源,笔者运用了密度泛函理论(DFT)。Ga3+未直接产生任何隙间态。此外,能带偏移的数值说明GaAs/HfO2界面是防止载流子注入并穿透氧化物层的一个良好的候选物。
《3. 方法》
3. 方法
为避免在周期性结构中顶部原子与底部原子直接出现相互作用,笔者使用了10Å的真空区域。底部的Ga原子用赝氢(带有1.25个价电子)进行了钝化处理,以模仿晶体内部的As键。在单胞中,HfO2的顶层最初由10个O原子终止;为了形成绝缘HfO2表面,10个O原子中的一半被移走了。在顶部(HfO2)表面和底部(GaAs)表面进行钝化能够保证消除顶部和底部表面态,进而保证所有计算出的隙间态源于界面。GaAs平板厚27.16Å,拥有10层Ga和9层As,而HfO2平板厚13.42Å,拥有5层Hf和6层O。此平板的厚度足以降低量子尺寸效应[36,37]。结构优化采用了共轭梯度(CG)法,且只固定了GaAs钝化层的底部。力收敛标准为0.01eV·Å–1。
界面电子态的性质在很大程度上取决于原子结构和界面键合的具体情况,这意味着准确的界面模型十分重要。在建造令人信服的HfO2/GaAs界面的理论模型过程中,必须考虑以下四个问题:第一,应变需适应晶格失配;第二,界面在理论上应当是稳定的和现实的;第三,应当考虑实验信息,且实验信息必须验证理论模型;第四,必须尽可能地降低搜索弛豫界面能量最小值的DFT限值。
在这项研究中,笔者考虑了立方HfO2与GaAs之间的界面。尽管HfO2存在立方相、四方晶相、单斜晶相,所有这些相都具有非常类似的局部离子键合特点,而且原子结构密切相关[38]。立方HfO2在原子结构优化期间可以转化为能量更低的结构。此外,当前分析所得出的结论也适用于其他相,因为核心要求在于价态的满足,这取决于局部键合结构,而非远程晶体对称性。笔者使用带有界面Ga—O(由Ga终止的GaAs表面和O终止的HfO2表面形成)的周期平板模型,这得到了实验数据的支持[39,40]。为了与GaAs(001)表面相匹配,(001)方向的HfO2结构被压缩了~0.3%,且逆时针旋转了28.04°(即28.04=45–16.96)(图6)。CG法[41]被用于原子结构优化。因为CG法[41]只能产生局部最小值,所以优化结构有可能是亚稳界面结构。为考察GaAs表面与HfO2表面之间形成的不同界面结构,在相对于GaAs平板的x方向和xy方向上移动HfO2平板,并将所获得的局部能量最小结构作为相对位移的函数。界面形成能在沿着xy方向上位移1.0Å 时减少了1.5eV,从而产生了最低总能量。笔者将这个最低能量界面结构作为初始结构,并进行全面弛豫。
《图6》
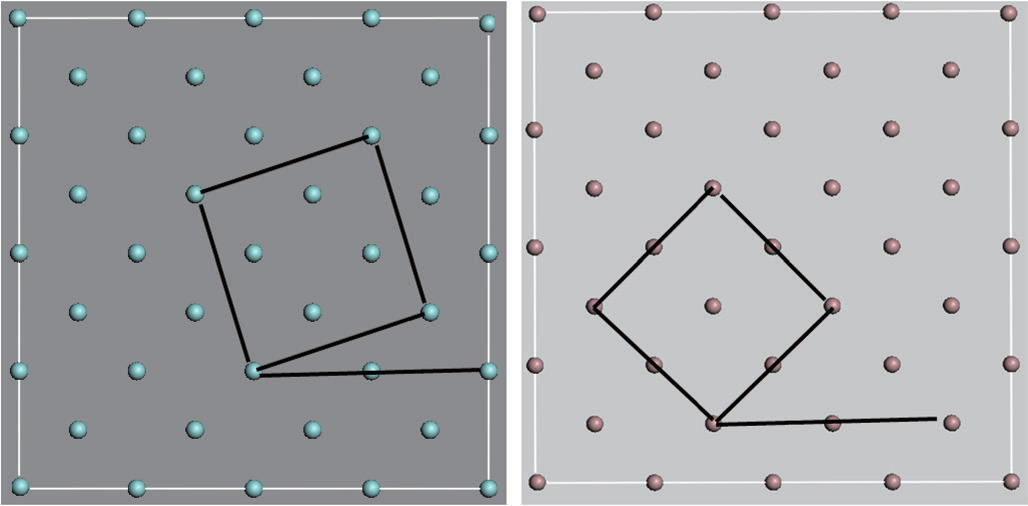
图 6. (a) 垂直于 (001) 方向的 HfO2 晶体内部的 Hf 单层;(b) 垂直于 (001) 方向的 GaAs (001) 晶体内部的 Ga 单层。翠绿色的原子代表 Hf,灰色的原子代表 Ga [20]。
在进行高能级电子结构计算时,为克服大家熟知的DFT限制[40,41]——带隙估计不足,笔者采用了HSE[42]计算。与局部密度近似(LDA)或广义梯度近似(GGA)[43,44]相比,HSE[42]产生的带隙和平衡晶格参数更符合实验结果。25%Hartree-Fock 交换势被纳入Perdew-Burke-Ernzerhof(PBE)势[45],所得带隙值为1.40eV;该结果足够接近实验值1.42eV[46]。
《致谢》
致谢
本研究得到了国家自然科学基金(11304161,11104148和51171082)、天津市自然科学基金(13JCYBJC41100和14JCZDJC37700)、国家重点基础研究发展计划(973项目)(2014CB931703)、高等学校博士学科点专项科研基金(20110031110034)和中央高校基本科研业务费专项资金的资助。Kyeongjae Cho得到了韩国首尔国立大学多尺度能量系统全球前沿中心的资助。笔者感谢美国奥斯汀市德克萨斯大学德克萨斯高级计算中心 (TACC)(http://www.tacc.utexas.edu) 在提供计算资源时的支持。
《Compliance with ethics guidelines 》
Compliance with ethics guidelines
Weichao Wang, Cheng Gong, Ka Xiong, Santosh K. C., Robert M. Wallace, and Kyeongjae Cho declare that they have no conflict of interest or financial conflicts to disclose.











 京公网安备 11010502051620号
京公网安备 11010502051620号




