
《1. 引言》
1. 引言
随着原油价格水平的提高,能够将煤、天然气和生物质转化为液态燃料和化学品的费托合成(FTS)技术,因可以使液态燃料来源与天然气、煤炭用途多元化,近年来引起了越来越多的关注[1,2]。费托合成的主要反应约90年前由Fischer和Tropsch发现[3]。费托合成反应通过强放热反应使一氧化碳(CO)加氢生成烷烃和烯烃,其方程式如下:

除烷烃和烯烃外,工业费托合成还可产生副产物含氧烃和二氧化碳(CO2)。费托合成反应的机理复杂,步骤繁多,诸如CO解离、碳加氢、CHx链增长,以及导致烃产物脱附和氧移除的加氢与脱氢反应[4]。许多反应步骤仍然存在争议,其中,最突出的是活性位点的本质和CO的解离方式,这与费托合成反应中单体生成的问题密切相关。从实际应用的角度来看,改进费托合成催化剂的主要目的是提高催化活性和目标产品(如长链烯烃)的选择性以及增加催化剂寿命。
在异相催化中,催化性能依赖于催化剂的电子结构[5],而可调参数是活性相组成、晶粒尺寸、晶体结构、晶体形态,以及过渡金属纳米粒子与催化剂载体之间的界面作用[6–11]。用于催化费托合成反应的典型过渡金属是钌(Ru)、钴(Co)和铁(Fe)。这些催化剂在低温费托合成反应中对液态烃均具有高活性和选择性[12]。尽管Ru的活性高,但价格昂贵,因此Co和Fe一直是工业费托合成中催化剂的主要成分。而基于镍(Ni)的催化剂在实际反应条件下通常产生过多的甲烷(CH4)[13]。本文我们将回顾通过费托合成工艺生产烷烃、烯烃和醇的Co、Fe和Ru催化剂领域的最新进展。文中将重点论述晶粒尺寸效应和晶相的影响,催化剂合成、现代表征技术和密度泛函理论计算在其中发挥更加重要的作用。本文包括三个主要部分:①讨论费托合成中Ru、Co和Fe催化剂的晶粒尺寸效应;②费托合成中Co、Ru、Fe和Ni金属催化剂的晶相结构效应描述;③结论与展望。
《2.费托合成中Co、Ru、Fe催化剂的晶粒尺寸效应》
2.费托合成中Co、Ru、Fe催化剂的晶粒尺寸效应
提高催化活性的一般方法是通过减小构成活性相的晶粒的尺寸来改善活性相的比表面积[7,11,14]。增加暴露的表面位置对非结构敏感的反应十分有效[11]。然而,许多催化反应是结构敏感的,这意味着特定的反应活性以更复杂的方式依赖于活性中心分散度[7,11,15–17]。当金属纳米粒子小于10nm时,这种效应尤其明显,因为诸如角位、边缘位、阶梯位的特定表面部位比一般晶面位在反应中更能起到决定性作用。由于费托合成反应是一种众所周知的结构敏感反应,因此许多研究者都专注于研究最佳粒径以获得高活性和选择性。鉴于尺寸效应在商业操作中的重要性,我们将仅讨论晶粒尺寸对Co、Ru和Fe纳米粒子催化剂的影响。
《2.1.费托合成Co催化剂的晶粒尺寸效应》
2.1.费托合成Co催化剂的晶粒尺寸效应
Iglesia[18]的研究表明,当以氧化铝(Al2O3)、二氧化硅(SiO2)或二氧化钛(TiO2)作为载体时,Co活性位点-时间产率在Co晶粒尺寸为10~210nm时与Co晶粒尺寸无关。Bezemer等[15]则认为当Co纳米晶粒(负载于碳纳米纤维)大于6nm时,CO加氢活性不敏感,但研究认为较小的Co纳米晶粒(<6nm)具有较低的CO加氢活性和较高的CH4选择性(图1)。稳态同位素瞬态动力学分析(SSITKA)测量表明,小Co纳米晶粒上较低的CO消耗速率可能是由于CO对边缘/角位点的阻塞所致[7]。小晶粒上的高氢覆盖率很好地解释了在小Co纳米晶粒上观察到的高CH4选择性。这种变化趋势也被其他研究组在SiO2负载的Co催化剂上证明[19,20]。对此现象的另一种解释是较小的Co纳米晶粒(<2.5nm)很容易被水氧化[19]。而这些关于晶粒尺寸效应的不同观点可能源于不同的反应条件。
《图1》
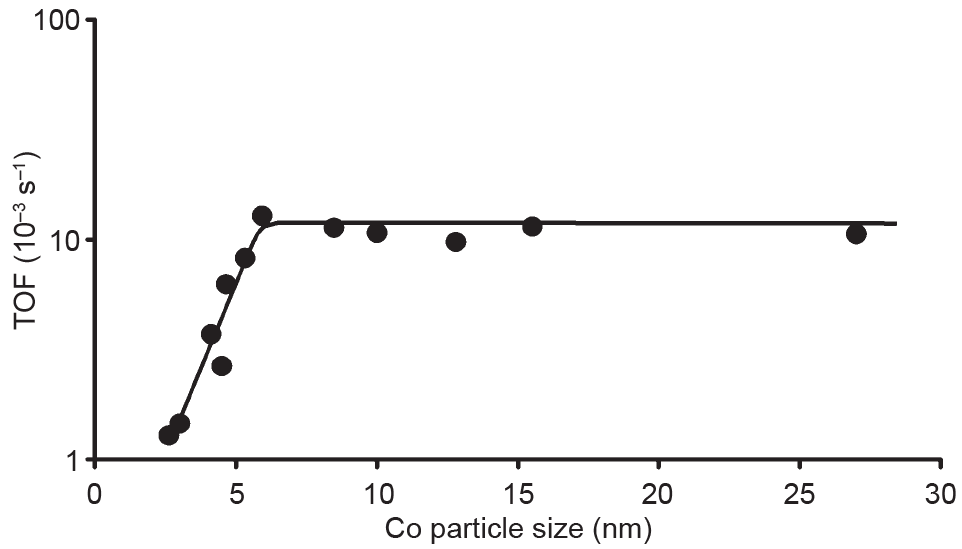
图1.费托合成中Co的晶粒尺寸效应[220℃,H2:CO=2:1,1bar(1bar=105Pa)][15]。TOF:转化频率。
Herranz等[21]的研究表明Co纳米晶粒在甲烷化条件下保持金属态。尽管当晶粒变得小于约10nm时,Co/SiO2催化剂的CO甲烷化活性(TOF)降低,但CO甲烷化的表观活化能对Co粒径并不敏感(图2)。氢氘(H-D)同位素置换实验表明,在小Co纳米晶粒上H2的解离很困难,这可以解释为较小晶粒上转化速率较低。而进一步的研究提供了证据,表明Co纳米晶粒上的解离氢对CO活化有促进作用,因而解离H2的能力是决定费托合成活性的关键参数[22]。然而,Holmen等[23]的研究提供了费托合成中Co尺寸效应的另一种观点。他们的SSITKA实验表明,表面CHx中间体的数量随着晶粒尺寸的增加而增加,而当Co晶粒尺寸从4nm变化到15nm时,本征活性保持恒定。他们的研究认为每个位点的活性在4~15nm不会发生变化,但是与大晶粒相比,小晶粒的活性位点相对较少。
《图2》
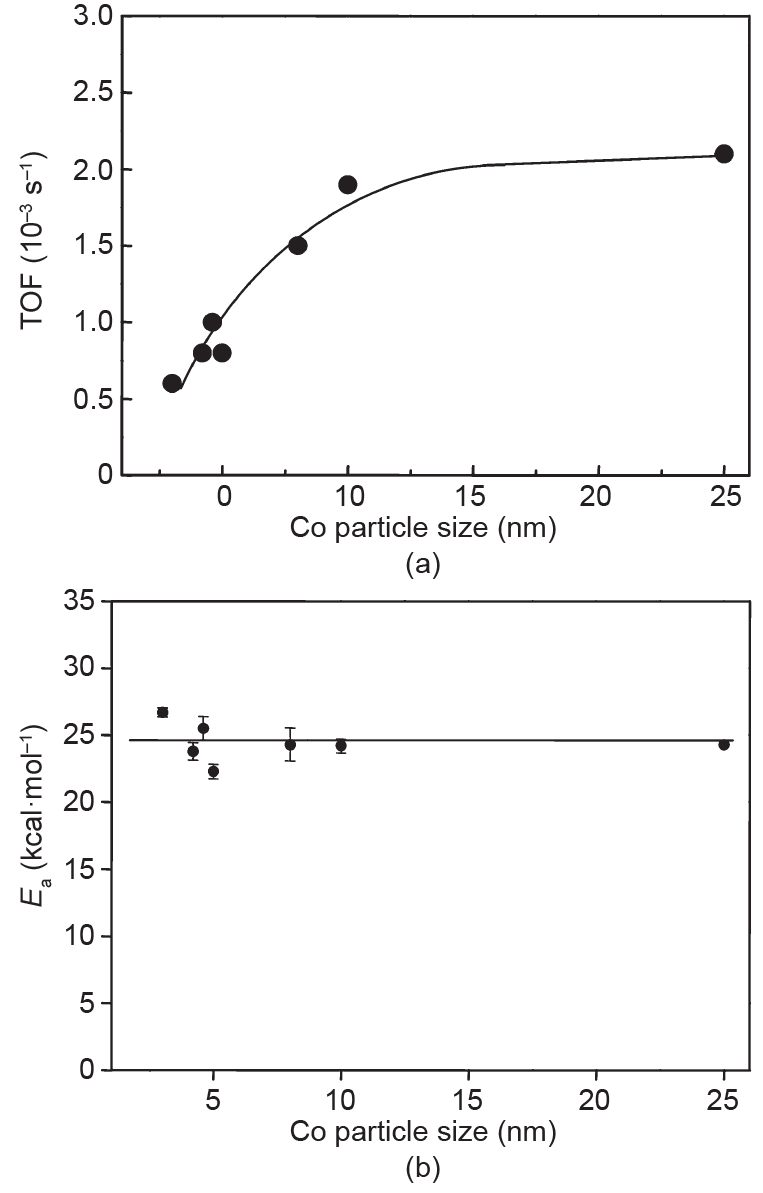
图2.CO加氢活性与Co晶粒尺寸的函数关系。(a)240℃下的CO加氢反应的TOF;(b)不同晶粒尺寸下CO加氢的活化能[21]。Ea:活化能(1cal=4.184J)。
尺寸效应对费托合成的选择性也有影响,小的Co纳米晶粒通常表现为CH4选择性上升。而C5+选择性随着Co/γ-Al2O3催化剂晶粒尺寸的变化呈现火山型曲线,Co纳米粒子的尺寸为7~8nm时,其选择性最高[24]。Holmen[25]的横向研究证实了这一发现,表明C5+选择性的确在低于8~9nm时随Co粒径的增加而增加,而Co晶粒尺寸更大时则接近恒定值。有趣的是,C5+选择性的最高点同时也是CH4选择性的最低点以及烯烷比的最高点。Melaet等[26]发现,在250℃条件下的产物分布中,费托合成反应随Co晶粒尺寸变化(3.2~11nm)而明显变化。图3显示CH4选择性随Co粒径减小而增加,同时C5+选择性降低。这一发现表明,大的Co纳米晶粒可以产生更长的烃产物链。然而,为何较大的Co纳米晶粒在费托合成中能够产生更长的烃产物链至今仍是一个开放问题。
《图3》

图3.CO加氢产物(C5+和CH4)选择性与Co晶粒尺寸的函数关系(250℃,H2:CO=2:1,5bar)。所有选择性的计算均基于转化的总碳原子数[26]。
《2.2.费托合成中Ru催化剂的晶粒尺寸效应》
2.2.费托合成中Ru催化剂的晶粒尺寸效应
DallaBetta等[27]和Iglesia等[28]的研究表明Ru催化剂的费托合成反应的本征活性并不强烈地依赖于晶粒尺寸,因而他们提出费托合成反应是非结构敏感的。然而,其他研究者的另外两项研究却得出了相反的结论,即费托合成反应活性是对Ru晶粒尺寸和金属分散度敏感的[17,29]。后者的发现得到了Kang[30]研究的支持,其研究表明费托合成反应的TOF和产物选择性强烈依赖于Ru催化剂的粒径。碳纳米管负载Ru催化剂在Ru晶粒尺寸约7nm时显示出对长链烃的最高活性和选择性。Xiao等[31]开发了用于水相费托合成的Ru纳米团簇催化剂。具有约2nm平均粒径的Ru催化剂在非常规的水相反应条件下具有极高的活性。具体地说,当晶粒尺寸减小到2nm时,费托合成活性显著增加,其中较小的团簇表现出较低的性能。然而,这些研究者没有明确解释Ru晶粒尺寸效应。我们的工作表明,小的无负载Ru纳米晶粒具有更低的费托合成活性,且在低温条件下水相费托合成表现出前所未有的70%的含氧化合物选择性[32–33]。
Carballo等[16]研究了具有不同金属粒径的Ru/Al2O3催化剂,并发现费托合成反应的TOF随Ru粒径的增加而增加,当Ru纳米粒子大于10nm时TOF趋于恒定。这与Co催化剂相似,如图4所示。此外,费托合成反应速率常数并不随Ru粒径而变化,这表明Ru催化剂上活性位点的本征活性并不受晶粒尺寸影响。笔者认为,小的Ru纳米粒子(<10nm)的较低TOF值可能与CO的强吸附有关。因此,较高的CO覆盖度将导致在较小的Ru纳米粒子上进行CO活化更加困难。此Ru催化剂尺寸效应规律与Xiao等[31]的水相费托合成反应研究并不相同。
《图4》

图4. CO消耗率 和CH4 生成率 与Ru晶粒尺寸的函数关 系(523 K; 5.5 kPa CO, 55 kPa H2, 124.5 kPa惰性气体)[16]。
以上关于Ru尺寸效应影响研究中的不同结论可能源于不同的论据。我们最近关于Ru催化剂水相费托合成反应的研究[33]表明,费托合成的TOF随着粒径的增加(1~5nm)而增加,高值是在2.3~3.7nm。小晶体团簇没有可以解离CO的阶梯位点,因而降低了费托合成活性。当晶粒大于2.5nm时,成烃的链增长参数不受其影响。
《2.3. 费托合成中 Fe催化剂的晶粒尺寸效应》
2.3. 费托合成中 Fe催化剂的晶粒尺寸效应
过去的实验表明,费托合成的FTS活性和选择性与Fe催化剂的粒度密切相关。例如,Mabaso等[34]提到,含有小于7~9nm的Fe纳米晶粒的催化剂具有比含有较大晶粒的催化剂更低的TOF和更高的CH4选择性。烯烃选择性不受晶粒尺寸影响,而小的Fe纳米晶粒显示出较低的链增长概率和较高的CH4选择性。这些发现被Liu等[35]的研究证实,他们的研究表明较小的Fe纳米晶粒会产生包括CH4在内的更多的短链烃。进一步的研究表明,当Fe晶粒尺寸从2.4nm增加到6.2nm时,TOF增加,之后晶粒尺寸增至11.5nm,TOF几乎保持恒定,如图5所示[36]。这组Fe催化剂的CH4选择性也随着晶粒尺寸的减小而上升。
《图5》
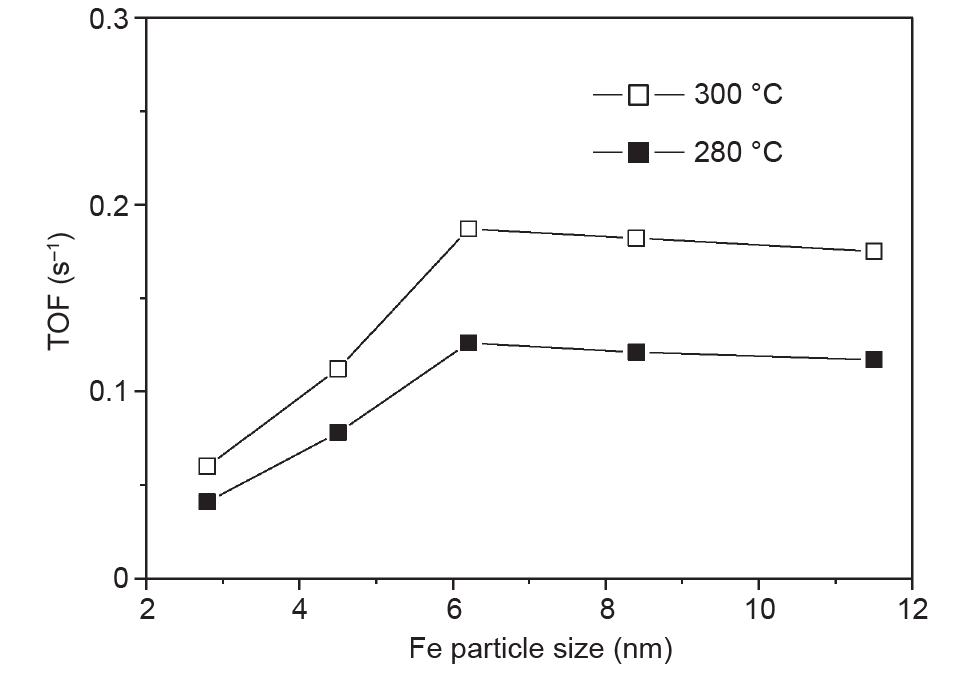
图5. 费托合成反应的TOF与Fe晶粒尺寸的函数关系。反应温度T = 280℃和300℃ [36]。
TorresGalvis等[12]研究了使用碳纳米纤维作为载体的碳化铁晶粒尺寸的影响。这些催化剂随着晶粒尺寸从2nm增加到7nm而呈现出活性降低趋势。然而,Fe催化剂的晶粒尺寸对烯烃或CH4选择性没有影响。与无助剂的碳化铁催化剂相反,加入Na和S助剂的催化剂显示出非常不同的催化行为。CH4和C2+烃表观TOF与晶粒尺寸的函数关系如图6所示[12]。研究发现当碳化铁粒径从7nm减小到2nm时,CH4选择性增加。然而,烯烃的选择性几乎与碳化铁晶粒尺寸无关。较小的碳化铁晶粒显示出较高的活性,其原因主要是较高的CH4产量。笔者提出,角位和边缘位是CH4形成的重要位点,而平台位则是可以生成烯烃的位点。晶粒尺寸的减小导致阶梯位和边缘位更加丰富,最终导致较高的CH4产率[12]。
《图6》

图6.CH4与C2+烃表观TOF随碳化铁尺寸变化的关系(反应时间=1h)。反应条件340℃,20bar,H2/CO=1,有助剂催化剂[12]。
上述结果表明,费托合成反应的催化活性与Co、Fe和Ru基催化剂的晶粒尺寸密切相关。虽然关于尺寸效应的作用原理仍然存在争议,但是对于如何合理设计具有最高特定反应活性的高效稳定催化剂,这些见解大有裨益。
《3.费托合成中催化剂金属粒子的晶相效应》
3.费托合成中催化剂金属粒子的晶相效应
除了尺寸效应外,研究也发现费托合成活性和选择性与催化剂的晶相关系紧密[37–56]。具有不同晶体结构的催化剂通常具有不同的形态和表面拓扑,由此暴露不同密度的活性位点,可导致不同的催化性能。在此,我们将聚焦近期关于Co、Ru、Fe和Ni催化剂的晶相效应研究。
《3.1.费托合成Co催化剂的晶相效应》
3.1.费托合成Co催化剂的晶相效应
对于Co催化剂,大多数研究者认为金属Co纳米晶粒是费托合成反应中的活性相。体相Co在正常环境条件下为六方最密堆积(HCP)结构。在400℃下,HCPCo与亚稳态面心立方(FCC)Co之间可发生温度诱导相变[57]。当Co的粒径非常小时,在更温和的条件下也可观察到这种HCP至FCC的转化。具体地说,当粒径在40nm以上时,HCPCo是主要的晶相,而当Co纳米粒子小于20nm时,则表现为FCC晶相[58]。在费托合成条件下,金属Co纳米粒子的实际晶体结构取决于载体类型、是否含助剂,以及晶粒尺寸[58]。催化剂的预处理/活化也将影响其结构[59–61]。这些晶体结构变化可能在费托合成活性和选择性中起重要作用。
据报道,HCPCo表现出比FCCCo更高的费托合成活性[37,44,45,50–52,62]。特别是Ducreux等[45]报道,主要由HCPCo纳米晶粒组成的Co催化剂具有比主要由FCCCo纳米晶粒组成的催化剂具备更高的费托合成活性[图7(a)]。而另一项工作表明,FCCCo纳米晶粒的活性低于HCPCo纳米晶粒;而碳化钴(Co2C)纳米晶粒对费托合成无活性[图7(b)][37]。近期,Davis与其同事[52]发现含有HCP相的Co催化剂在费托合成中显示出比FCCCo纳米晶粒更高的CO转化率。与FCCCo纳米晶粒相比,HCPCo纳米晶粒在C2~C4烃范围内表现出略高的烯烃选择性,且CH4的生成速率较低。研究者提出CO在HCPCo纳米晶粒上的高转化率可能是由表面缺陷和堆垛层错密度较高造成的[45,51]。
《图7》

图7.(a)Co-Ru/SiO2催化剂上CO转化率与时间的函数关系:(i)FCC-HCP混合结构(菱形图标);(ii)FCC结构(正方形图标);(iii)HCP(三角形图标)[45]。(b)在FCCCo、HCPCo和Co2C催化剂上的Co活性位点费托合成产率[37]。
尽管有众多研究,但为何HCPCo纳米晶粒比FCCCo纳米晶粒活性更高仍然是一个悬而未决的问题。Liu等[39]通过第一性原理密度泛函理论(DFT)计算比较了HCPCo和FCCCo纳米晶粒上的CO活化。HCPCo和FCCCo纳米粒子的形貌可以近似用晶体学中的Wulff构建原理确定[63]。如图8所示,HCPCo和FCCCo纳米晶粒呈现不同的形态。基于计算得到的CO活化能垒,可通过渡态理论估算CO解离速率。这项研究的一个主要结论是HCPCo纳米晶粒的四个晶面,即 和
和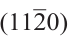 ,表现出比FCCCo纳米晶粒中活性最高的FCC(100)晶面更高的CO解离速率。HCPCo纳米晶粒显示出比FCCCo纳米晶粒更高的CO解离活性,因为存在更多的高活性B5位点。该工作进一步证实,CO活化反应途径对于HCPCo和FCCCo相也是不同的,即CO倾向于在HCPCo相上直接解离,而在FCCCo相上H辅助路线则更重要。然而即使考虑H辅助CO活化途径,HCPCo相亦显示出比FCCCo相更高的活性。
,表现出比FCCCo纳米晶粒中活性最高的FCC(100)晶面更高的CO解离速率。HCPCo纳米晶粒显示出比FCCCo纳米晶粒更高的CO解离活性,因为存在更多的高活性B5位点。该工作进一步证实,CO活化反应途径对于HCPCo和FCCCo相也是不同的,即CO倾向于在HCPCo相上直接解离,而在FCCCo相上H辅助路线则更重要。然而即使考虑H辅助CO活化途径,HCPCo相亦显示出比FCCCo相更高的活性。
《图8》
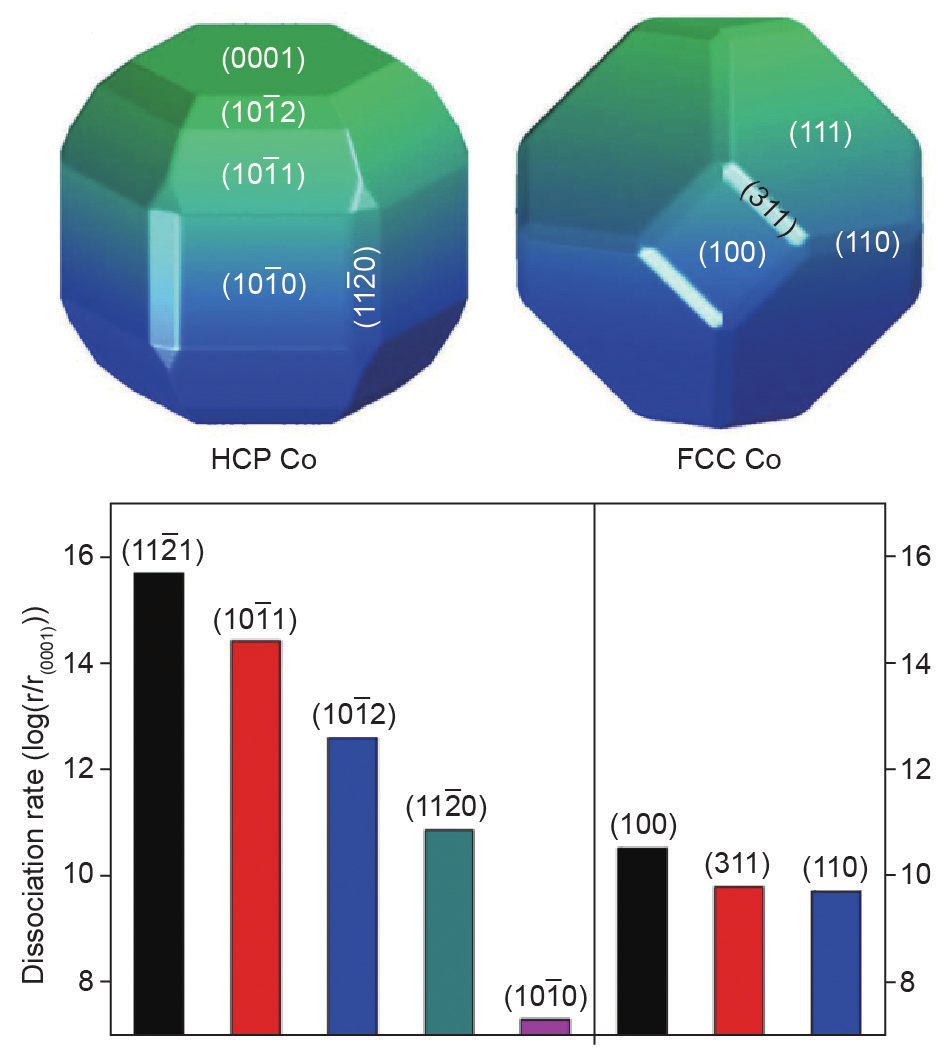
图8.HCPCo与FCCCo纳米晶粒的Wulff构建及HCPCo与FCCCo纳米晶粒不同晶面上CO的解离速率。所有速率均归一化为HCP(0001)晶面上的速率[39].
《图9》
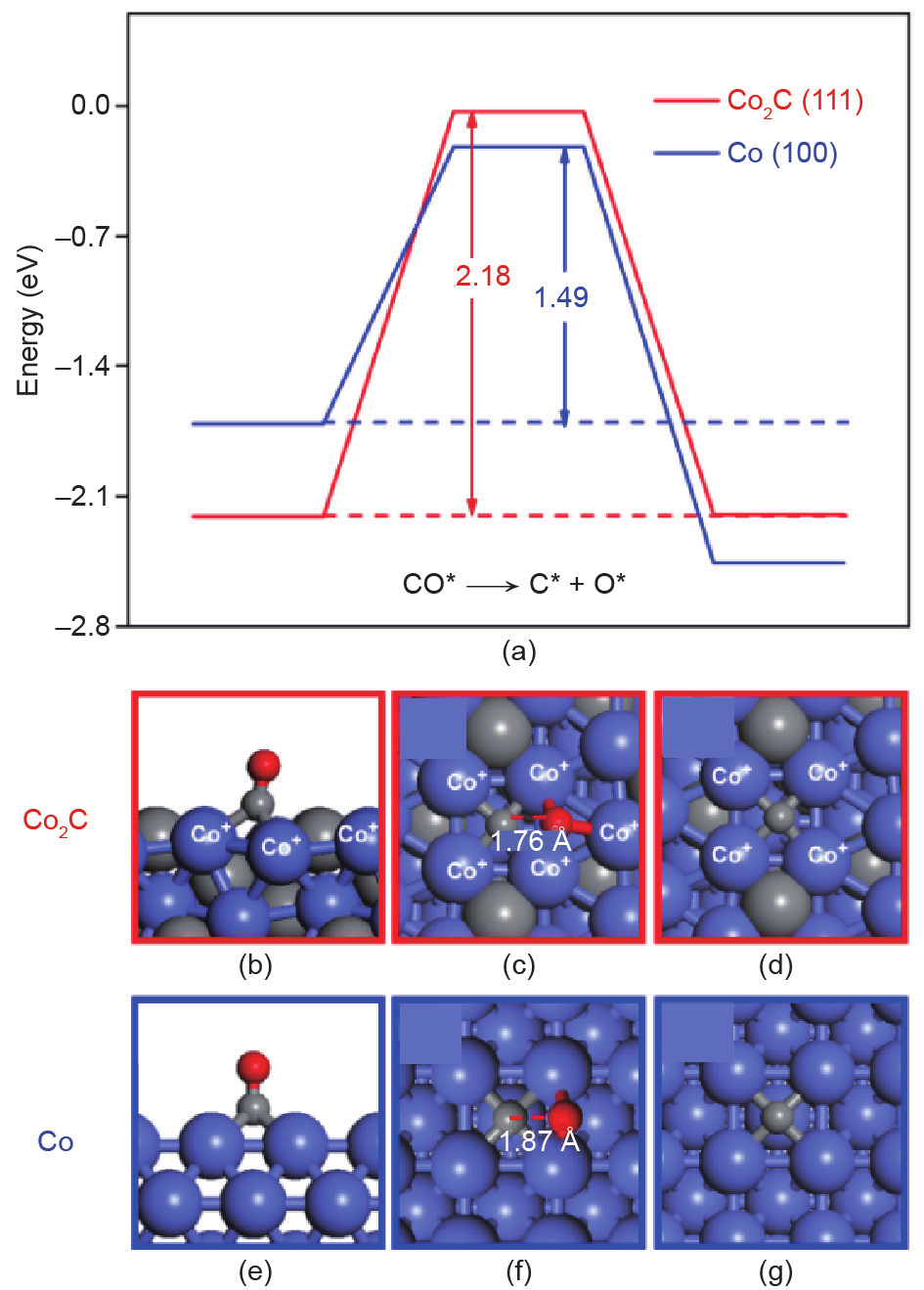
图9.(a)晶面势能与(b~g)CO在Co2C(111)(红色)和Co(100)(蓝色)晶面的活化形态[55]。
最近,Zhong等[53]合成了表现出高费托合成活性的含锰(Mn)助剂的Co2C纳米棱柱。在温和的费托合成反应条件下,这种Co2C催化剂在相对低的CH4选择性下能产生更多的轻质烯烃(C2~C4烃)(图10)。此外,Co2C纳米棱柱在前15h显示出优异的稳定性。这个结果非常令人惊讶,因为传统的球形Co2C纳米晶体对于费托合成显示出极低的活性。透射电子显微镜(TEM)表征和DFT计算显示,优先暴露的(101)和(020)面可能是合成气转化为烯烃的特定活性位点。这项工作为设计下一代高效的费托合成制烯烃(FTO)催化剂提供了新的途径。
《图10》
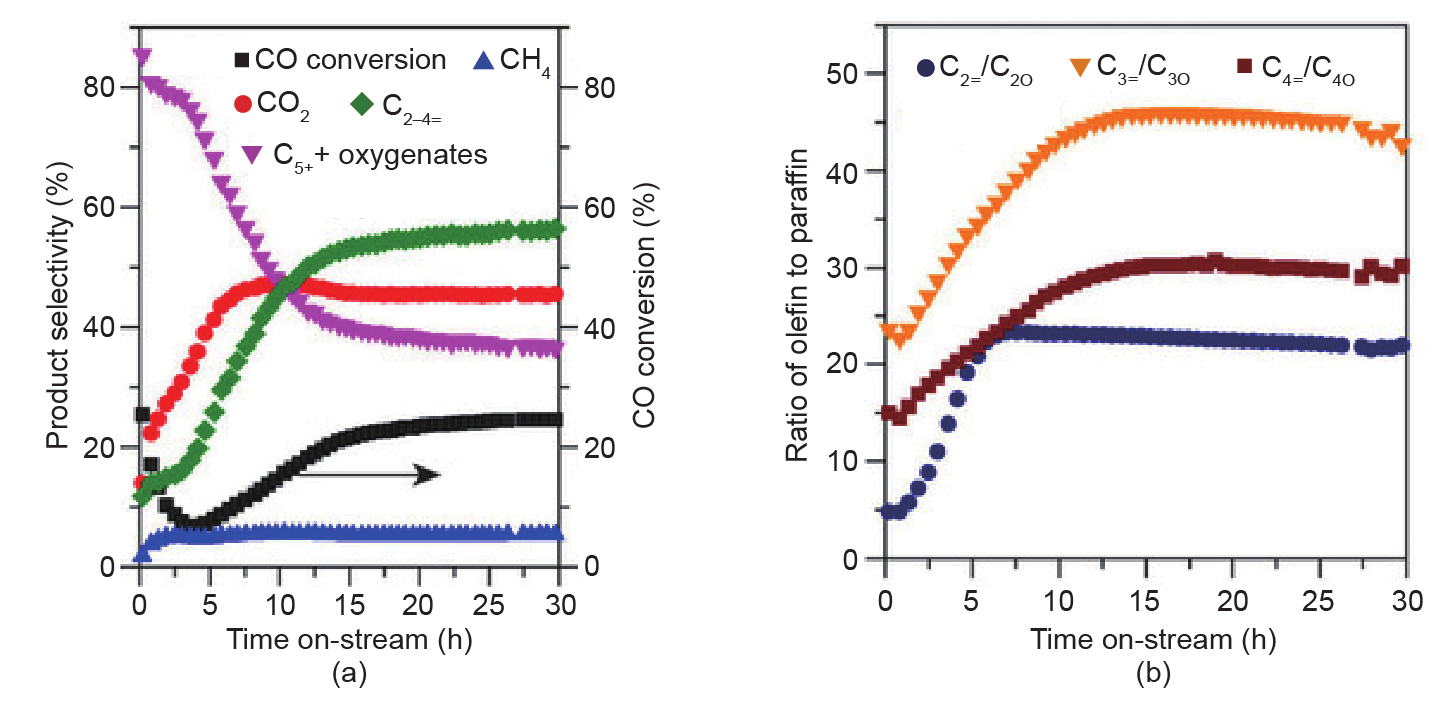
图10. CoMn催化剂在反应初始阶段的催化性能。(a)CO转化率和产物选择性与反应时间的函数关系;(b)产物烯烷比与反应时间的函数关系[53]。
《3.2.费托合成Ru催化剂的晶相效应》
3.2.费托合成Ru催化剂的晶相效应
Ru纳米粒子广泛用于异相催化。研究者对Ru催化剂的晶体结构与其性能之间的关系一直有着浓厚的兴趣。Kusada等[40]合成了纯FCCRu纳米晶粒催化剂。他们发现CO氧化活性强烈依赖于Ru催化剂的晶体结构和粒径。当晶粒大于3nm,传统的HCPRu纳米晶粒的活性低于FCCRu纳米粒子。研究者提出FCCRu纳米粒子的较高活性源于大量存在的Ru(111)晶面很容易被氧化为RuO2(110),而RuO2(110)比金属Ru更具CO氧化活性。后来,Gu等[42]合成了具有FCC结构并且被Ru(111)面大部分覆盖的Ru@Pt核-壳结构催化剂。这种Ru@Pt核-壳结构催化剂比常规HCPRu催化剂对析氢反应更有活性[42]。在其他反应中也观察到FCCRu纳米晶粒比HCPRu纳米晶粒有更高的活性,如氨硼烷转化[48,68]、析氧反应[69]、加氢反应[70]和氮气活化反应[71]。因此,关于Ru催化剂的FCC和HCP晶体结构如何影响费托合成的活性和选择性的问题仍然是一个有趣和极具时效性的论题。
Li等[72]近期通过DFT计算、现代材料合成和扫描透射电子显微镜(STEM)的组合,研究了FCC和HCP结构的Ru催化剂费托合成。DFT计算表明,在FCCRu纳米晶粒催化剂(图11)上有许多适于CO解离的低能垒开放晶面,但在HCPRu纳米晶粒催化剂上则只有一些阶梯边缘位点上能垒较低。基于这些理论结果,研究者合成了具有高密度活性位点(开放晶面)的FCCPt@Ru核-壳结构纳米晶体催化剂。合成的FCCRu纳米晶粒催化剂在393~433K的低温范围内显示出非常高的水相费托合成活性。费托合成的活性在433K时高达37.8molCO∙mol−1Ru∙h−1,使其成为迄今为止在低温(<473K)下活性最高的费托合成催化剂。与HCPRu纳米粒子相比,FCCRu纳米粒子的高活性源于较高的活性位点密度。值得注意的是,HCP和FCC相之间的催化行为趋势差异取决于过渡金属本身。例如,FCCCo纳米晶粒的活性低于HCPCo纳米晶粒,而Ru纳米晶粒显示出相反的趋势。Ru和Co纳米晶粒在费托合成中的动力学差异主要源于其结构和电子差异,而这些差异又源于Ru纳米晶粒的较大的晶格常数。
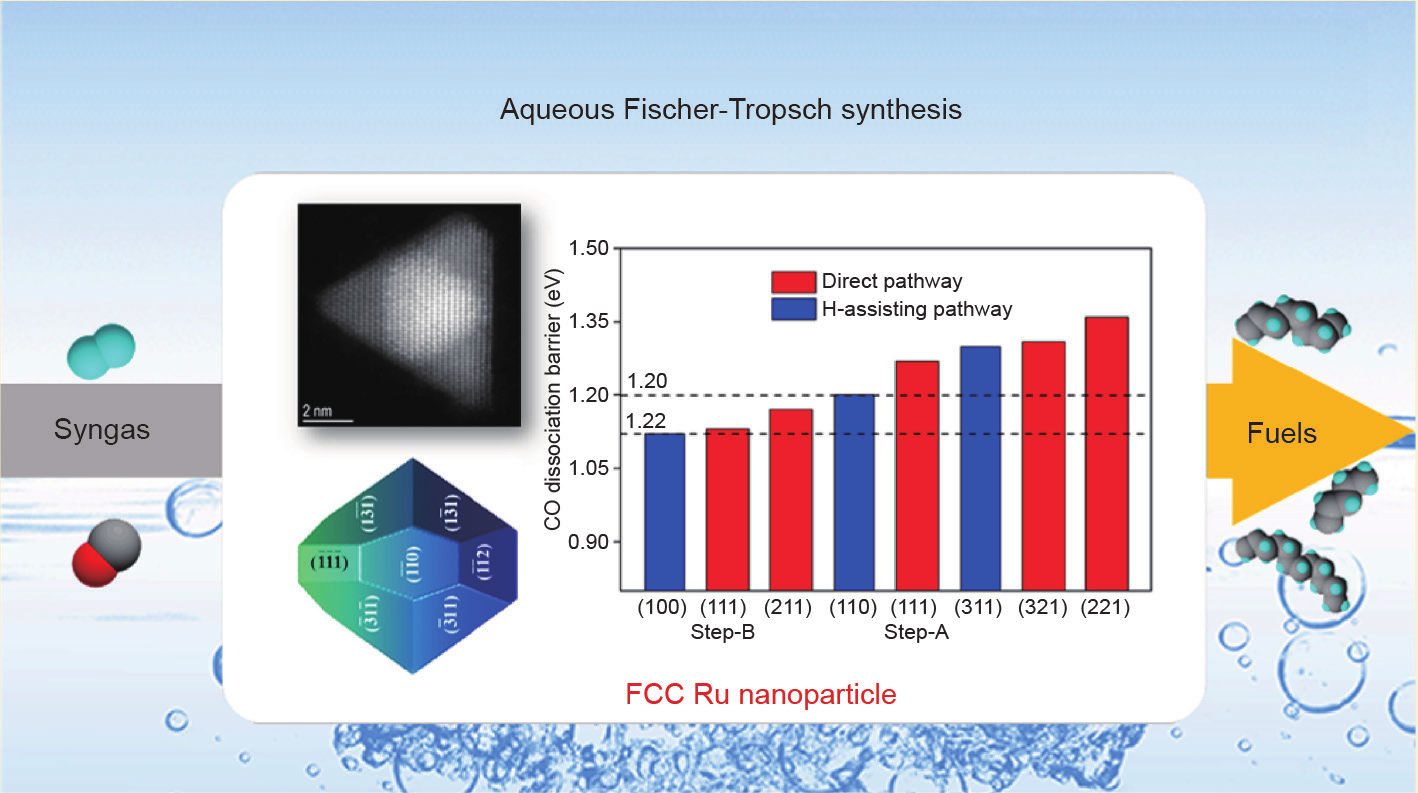
图11. FCC Ru纳米晶粒上各晶面的CO活化能垒示意图。Wulff构建与STEM图像基于Pt@Ru纳米晶粒建模[72]。
《3.3. 费托合成 Fe 催化剂的晶相效应》
3.3. 费托合成 Fe 催化剂的晶相效应
合成纯相的碳化铁很难。Fe催化剂的活性相的变化非常复杂,这导致其活性相本质和Fe催化剂的费托合成反应机理存在着相当大的争论。而了解不同相的分布、相变及其与活性的关系将有助于设计出最优的费托合成Fe催化剂。在费托合成反应条件下已观察到多种不同的碳化铁,包括ε-Fe2C、ε′-Fe2.2C、Fe7C3、χ-Fe5C2与θ-Fe3C。如图12所示,碳化铁相变的发生(ε-χ-θ相变)依赖于温度和H2/CO比例[73]。具体地说,高温和碳的低化学势(µC)即高H2/CO比例,通常导致θ-Fe3C的优先形成。相反,高µC(低H2/CO比例)和中等温度(约250℃)则导向χ-Fe5C2的形成。ε-碳化物则是优先在较低温度、更高的µC下形成。
《图12》
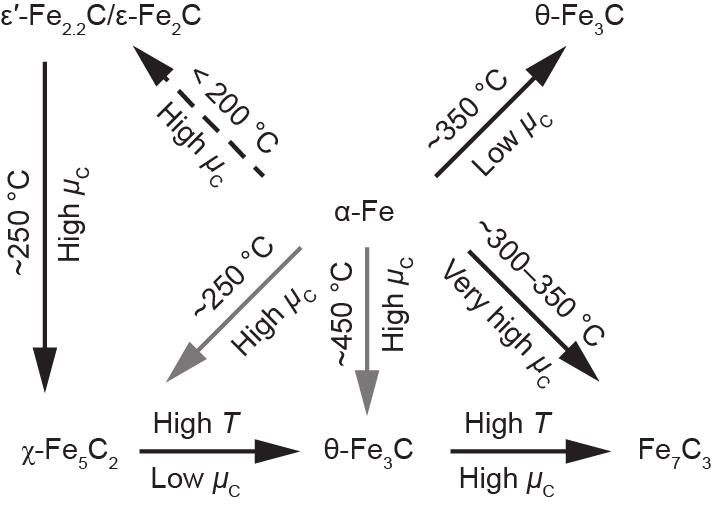
图12. 碳化铁物相随温度(T)与碳化学势的变化[73]。
辨识费托合成的碳化铁活性相在异相催化中仍然是一个很大的挑战。据称金属Fe[74,75]和各种碳化铁相均被认为对费托合成反应是有活性的[76–80]。DeSmit等[73]报道,Fe催化剂主要由χ-Fe5C2组成,它具有费托活性但易被氧化。与此相比,θ-Fe3C催化剂则显示出较低的活性和选择性,这可能是由于催化剂表面上的沉积碳质积累所致。Fierro的实验证实了这一发现,表明χ-Fe5C2比θ-Fe3C催化剂更具费托合成活性[81]。随后,Yang等[82]首先报道了液相化学法合成纯χ-Fe5C2纳米粒子的方法。这些χ-Fe5C2纳米粒子催化剂具有比传统的还原赤铁矿催化剂更高的活性和选择性。
《图13》

图13. 费托合成Fe催化剂在不同反应条件下的形态[83]。
近期,Yang等[84]报道了高费托合成活性的二氧化铈(CeO2)负载的亚纳米氧化铁簇催化剂。研究利用STEM和X射线吸收精细结构谱(XAFS)表征,证实CeO2纳米棒上负载的氧化铁簇(Fe-Ox-Fey)中含有的部分还原的Fed+(d=2.6~2.9)为费托合成活性相。有趣的是,这些研究人员在费托合成反应期间没有观察到这种催化剂的任何铁碳化物。他们的工作表明,费托合成活性与活性位点局域的配位状况密切相关,碳化铁并不一定是费托合成的必须活性相。虽然研究众多,但仍需进行更多的实验和理论工作才能明确在实际条件下费托合成Fe催化剂的真实活性相。
《3.4.费托合成Ni催化剂的晶相效应》
3.4.费托合成Ni催化剂的晶相效应
Ni不是一个很好的费托合成催化剂,因其较差的CO解离的能力,使其主要被用在制CH4反应中,且其稳定性也不好,这是由于其在实际的费托合成反应条件下易形成挥发性羰基[85]。然而,Ni可用作生产CH4的有效催化剂,也可用作费托合成的助剂[86]。体相Ni在通常条件下为FCC结构。然而,当Ni晶粒尺寸减小到4nm时,FCCNi可以转化为HCPNi[87]。事实上,HCPNi纳米晶粒已可通过一步化学途径[87]或其他化学方法[88–95]合成。研究发现在甘油的水相重整中,HCPNi具有比FCCNi更高的活性。此外,与FCCNi纳米晶粒相比,HCPNi纳米晶粒可以呈现更高的H2选择性并阻碍CH4生成[96]。
CO活化是CO甲烷化反应的第一个关键步骤。有研究表明CO活化与Ni纳米晶粒的表面结构密切相关,而且不规则的阶梯/边缘位点是CO解离的活性位点[97,98]。通过COH中间体的CO活化被认为是CO甲烷化反应的决速步骤[99,100]。Liu等[54]通过DFT计算研究Ni晶体相对CO活化的影响(图14)。他们的计算分析表明,CO解离强烈依赖于Ni催化剂的晶体结构和形态。需要注意的是,不论哪种晶体相,由氢辅助的CO解离在动力学上均优于直接解离途径。如图14所示,(311)与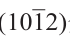 分别为FCC Ni和HCP Ni的最高活性晶面。CO解离更倾向于通过COH中间体在FCC和HCP Ni催化剂的最高活性晶面上实现。FCC Ni比HCP Ni更活跃,因为FCC Ni大量暴露出的晶面活化能垒较低。不同晶相(FCC、HCP)的不同反应行为揭示了很有价值的催化剂优化方向,即暴露更多活性位点有利于双原子分子的活化。
分别为FCC Ni和HCP Ni的最高活性晶面。CO解离更倾向于通过COH中间体在FCC和HCP Ni催化剂的最高活性晶面上实现。FCC Ni比HCP Ni更活跃,因为FCC Ni大量暴露出的晶面活化能垒较低。不同晶相(FCC、HCP)的不同反应行为揭示了很有价值的催化剂优化方向,即暴露更多活性位点有利于双原子分子的活化。
《图14》
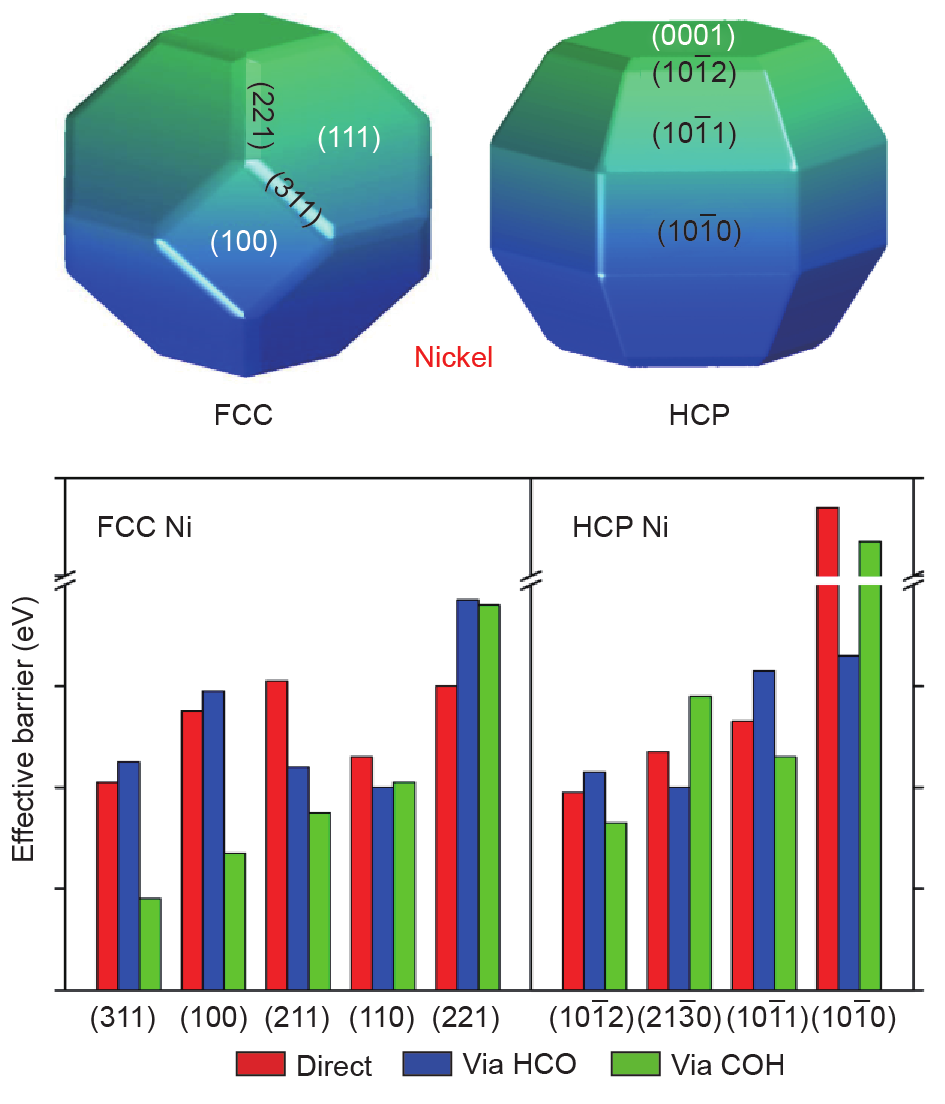
图14. 在FCC Ni与HCP Ni不同晶面上的CO活化反应,依据Wulff构建原理[54]。
《4. 结论与展望》
研究催化剂晶相结构与费托合成催化活性之间的关系将非常有助于最优催化剂设计。在本文中,我们总结了费托合成性能的晶粒尺寸效应和晶相效应的最新研究进展。费托合成活性和选择性强烈依赖于催化剂的晶粒尺寸。然而,由于费托合成反应机理的复杂性和原位表征的限制,费托合成晶粒尺寸效应的作用原因仍然存在争议。不同晶相和结构的催化剂具有不同的形态;所致使的不同的电子状态导致活性和选择性变化。鉴于此,开发现代表征技术和先进的材料合成方法,将其与DFT计算结合应用于模型催化剂和实际催化剂系统,以明确活性位点本质,探索能显著提高CO加氢生产化学品和燃料的金属催化剂性能的新理念,比以往任何时候都更加必要。
《Acknowledgements》
Acknowledgements
We acknowledge financial support by NWO-VICI and NWO-TOP grants awarded to Emiel J. M. Hensen.
《Compliance with ethics guidelines》
Compliance with ethics guidelines
Jin-Xun Liu, Peng Wang, Wayne Xu, and Emiel J. M. Hensen declare that they have no conflict of interest or fi- nancial conflicts to disclose.











 京公网安备 11010502051620号
京公网安备 11010502051620号




