
《1. 引言》
1. 引言
癌症是一项全球性公共健康问题,每年有数万人死于癌症。尽管科研人员在开发有效治疗策略方面做了很大努力,但结果不尽如人意,且大多数癌症的总生存率和无疾病生存期均很低。虽然放疗、化疗、外科手术和分子靶向治疗等传统治疗可有效攻击肿瘤细胞,但它们同时也会产生危及生命的不良作用。最常见的不良反应包括胃肠道反应和骨髓抑制,不仅会影响术后生活质量,还可直接导致死亡[1]。过去10年来,免疫疗法一直是肿瘤治疗的活跃研究领域,并且研究人员在这一领域付出了巨大努力。以免疫检查点抑制剂[如程序性死亡受体1(PD-1)、程序性死亡配体1(PD-L1)和细胞毒性T淋巴细胞相关抗原4(cytotoxic T lymphocyte-associated antigen 4,CTLA-4)与嵌合抗原受体T细胞免疫疗法(chimeric antigen receptor T cell immunotherapy,CAR-T)]为代表的新策略在解决各类癌症方面已显示出令人鼓舞的功效[2−8]。遗憾的是,越来越多的研究表明仅有20%~30%的患者在采用这些策略后出现缓解[9],于是便引发了为什么免疫疗法对其余70%~80%的患者无效的疑问。人们已经接受的一种解释是:肿瘤已形成了一个可防止免疫系统攻击的免疫抑制网络,调节性T细胞(Treg)是该网络的一个重要组成部分。高表达CD25[白细胞介素2(IL-2)受体α链]和FOXP3的调节性T细胞在肿瘤微环境中富集,是免疫抑制网络中的主要构成部分[10]。在本文中,我们重点关注调节性T细胞在肿瘤免疫抑制网络中发挥的重要作用,以及其在治疗多种肿瘤方面的治疗潜力。
《2. 调节性 T 细胞的分化与发育》
2. 调节性 T 细胞的分化与发育
根据来源,调节性T细胞可分为两大类。第一类是自然调节性T细胞(nTreg),其从胸腺分化且由广谱自身抗原产生[11];因此,自然调节性T细胞也被称为胸腺源性调节性T细胞(tTreg)。在受到主要组织相容性复合体(major histocompatibility complex,MHC)-自身抗原复合体的刺激后,胸腺细胞中的一部分CD4+ CD+T细胞开始表达CD25,与CD25结合后,IL-2可通过信号转导及转录激活蛋白5(signal transducer and activator of transcription 5,STAT5)诱发Foxp3的产生[12]。另一类为诱导性调节性T细胞(iTreg),也称为外周调节性T细胞(pTreg)。许多因子[如转化生长因子β(TGF-β)、表达吲哚胺2,3-双加氧酶(indole amine 2,3-dioxygenase,IDO)的树突细胞或视黄酸]均可将外周CD4+ 初始T细胞转化为诱导性调节性T细胞[13−15]。两大亚群有相似的表面标志物,且对效应T细胞(Teff)有相当的抑制作用。然而,不断有证据表明调节性T细胞亚群之间存在许多不同,如不同的蛋白质表达与表观遗传修饰。例如,据报道,诱导性调节性T细胞表达低水平的Helios与神经纤毛蛋白-1(NRP1),而自然调节性T细胞大量存在于这些分子中[16,17]。此外,还有另外两种不表达Foxp3的特殊调节性T细胞亚群,即IL-10产生性1型调节性T细胞(Tr1)和TGF-β产生性3型辅助性T细胞(Th3)[18]。
在调节性T细胞发育过程中,T细胞受体(T cell receptor,TCR)信号不可缺少,因为TCR阻断会导致调节性T细胞发育受到抑制[19]。对自身抗原有高亲和力的T细胞在胸腺中会经历阴性选择,导致细胞凋亡,而与自身抗原亲和力不高的T细胞将存活并发育成效应T细胞。选择用来产生自然调节性T细胞的亲和力阈值介于阳性与阴性选择之间[20]。此外,调节性T细胞发育需要共刺激分子,如CD28和糖皮质激素诱导的肿瘤坏死因子受体(tumor necrosis factor receptor,TNFR)相关蛋白(GITR)。之前的研究表明,在缺乏CD28或CD80/CD86的小鼠中,调节性T细胞的数量减少[21,22]。对调节性T细胞发育而言不可缺少的另一个因子是IL-2。调节性T细胞对高剂量IL-2、固体包衣或可溶性抗CD3单克隆抗体(mAb)或抗CD3单克隆抗体与抗CD28单克隆抗体的组合物均无反应。只有当TCR信号与高浓度外源IL-2同时呈递时,才能激活调节性T细胞。然而,其增殖却远远弱于CD4 + CD25 – T细胞的增殖[23]。事实上,抗原激活T细胞产生IL-2,以诱导调节性T细胞的增殖,而IL-2随后又增加对这些已激活T细胞的抑制性反应,以避免过度反应,从而构成一个负反馈的回路。TCR/CD28的下游即核因子κB(NF-κB)信号通路对调节性T细胞的发育也起着至关重要的作用。在TCR/CD28共刺激的情况下,c-REL与启动子和保守的非编码DNA序列结合以调控Foxp3转录[24]。髓质上皮细胞(medullary thymic epithelial cell,mTEC)信号也是调节自然调节性T细胞发育的一个重要因子。TNFR相关因子6(TN-FR-associated factor 6,TRAF6)缺陷损害髓质上皮细胞的成熟,并因此抑制调节性T细胞的发育[25]。在小鼠癌症模型中,研究者发现神经纤毛蛋白-1在维持肿瘤浸润调节性T细胞功能中起重要作用。选择性敲除小鼠的 将导致调节性T细胞功能缺失。这种表型的潜在机制是因缺陷而产生的干扰素-γ(interferon-γ,IFN-γ)增强了肿瘤浸润调节性T细胞的脆性[26]。值得注意的是缺乏将仅仅影响调节性T细胞的抗肿瘤功能,而不会影响其防止自身免疫疾病的作用。
将导致调节性T细胞功能缺失。这种表型的潜在机制是因缺陷而产生的干扰素-γ(interferon-γ,IFN-γ)增强了肿瘤浸润调节性T细胞的脆性[26]。值得注意的是缺乏将仅仅影响调节性T细胞的抗肿瘤功能,而不会影响其防止自身免疫疾病的作用。
《3. 调节性 T 细胞的功能调控》
3. 调节性 T 细胞的功能调控
FOXP3是调节性T细胞发育和功能的主调节因子[27]。在Foxp3敲除小鼠中,自然调节性T细胞明显减少。此外,Foxp3的过继转移可将外周血CD4+ CD25– T细胞转化为诱导性调节性T细胞。这两种研究结果均表明Foxp3是调节性T细胞发育的必要条件。Foxp3表达受到一系列生理信号和蛋白修饰调控。然而,除了调节性T细胞之外,还有多种表达Foxp3但缺乏抑制功能的细胞种类,包括乳腺、肺及前列腺上皮细胞[28]。这表明Foxp3表达对于建立调节性T细胞谱系而言是不够的。而且,在某些情况下,FOXP3– 细胞也拥有强大的抑制功能[29]。越来越多的证据表明在调节性T细胞的发育过程中存在两种独立活动:Foxp3表达和Foxp3修饰[30]。
《3.1. Foxp3 转录水平调控》
3.1. Foxp3 转录水平调控
Foxp3基因表达由四大元素控制:启动子区;第一内含子内的两个保守非编码序列(CNS1和CNS2)以及包括CNS3的第二内含子[31]。CNS1包含活化蛋白-1(AP-1)、SMAD3和活化T细胞核因子(nuclear factor of activated T,NFAT)的结合位点。在CNS1敲除小鼠中,调节性T细胞中Foxp3的表达与野生型(wild-type,WT)小鼠相当,而诱导性调节性T细胞有所减少,这一结果揭示了CNS1在调节性T细胞外周诱导中的作用。环孢素A(NFAT的抑制剂)和SIS3(SMAD3的抑制剂)可导致Foxp3表达缺失[32]。CNS2包含STAT5[33]、环腺苷酸(cAMP)应答元件结合(CREB)/活化转录因子(activating transcription factor,ATF)[34]、叉头框蛋白O1(forkhead box protein O1,FOXO1)和叉头框蛋白O3(FOXO3)[35]的结合位点,被称为“调节性T细胞特异性去甲基化区”(treg-specific demethylated region,TSDR),原因在于自然调节性T细胞中的该区域被完全去甲基化。CNS2缺失小鼠在6个月之后调节性T细胞减少,这表明CNS2对维持稳定性而言至关重要,但对Foxp3表达而言却是可有可无的[32]。CNS3受C-REL约束,C-REL作为染色质开端促进Foxp3特异性的增强体的形成;CNS3– /– 小鼠中的调节性T细胞数量减少[24]。
《3.2. 调节性 T 细胞中的 Foxp3 后修饰》
3.2. 调节性 T 细胞中的 Foxp3 后修饰
调节性T细胞中的Foxp3后修饰由泛素化、乙酰化、甲基化等构成。在由促炎性细胞因子和脂多糖引发的炎症下,调节性T细胞将下调Foxp3表达,由此使类似效应T细胞的功能增加[36]。这一表型的潜在机制如下:在炎症期间,活化E3泛素连接酶Stub1,Stub1诱导FOXP3以热休克蛋白70依赖方式降解。相反,泛素特异性蛋白酶7(ubiquitin-specific protease 7,USP7)和泛素特异性蛋白酶21(USP21)是能够减少调节性T细胞数量和功能的两大主要去泛素酶[37,38]。
赖氨酸乙酰化与去乙酰化之间的平衡是调节FOXP3蛋白稳定性和功能的另一重要因子。组蛋白乙酰转移酶(histone acetyltransferase,HAT)TIP60 [39]与p300 [40]可稳定FOXP3蛋白,并由此通过FOXP3乙酰化促进其抑制功能。这里,烟酰胺腺嘌呤二核苷酸(nicotinamide adenine dinucleotide,NAD)依赖性去乙酰化酶sirtuin-1(SIRT1)的作用与TIP60和p300的作用恰恰相反[41]。与传统T细胞相比,CNS2和Foxp3启动子含有对维持Foxp3表达不可缺少的高度去甲基化区[42]。第一内含子内CREB/ATF位点的甲基化表明其与Foxp3表达呈负相关关系。还存在许多其他调节方法,如磷酸化与小泛素样修饰因子(small ubiquitin-like modifier,SUMO)修饰。这是一个由多个元件调控的复杂过程。
《4. 调节性 T 细胞的功能与抑制机制》
4. 调节性 T 细胞的功能与抑制机制
《4.1. 调节性 T 细胞的抑制机制》
4.1. 调节性 T 细胞的抑制机制
调节性T细胞通过多种机制发挥其抑制活性(图1)。以下列出了其中的一些机制。
《图1》
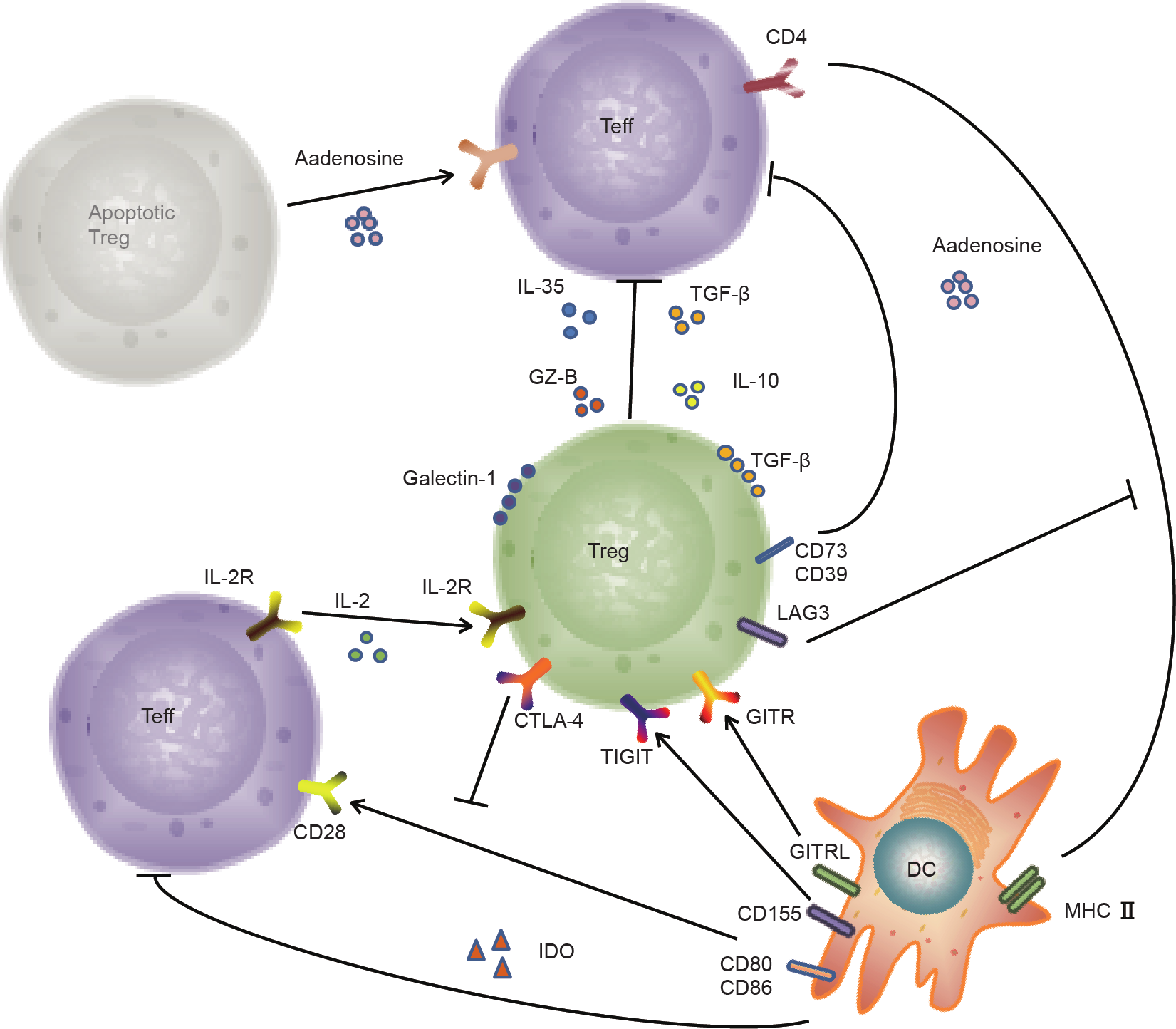
图1. 调节性T细胞的抑制机制。GITR:糖皮质激素诱导的肿瘤坏死因子受体相关蛋白;GITRL:GITR配体;LAG3:淋巴细胞活化基因3;GZ-B:颗粒酶B;MHC II:主要组织相容性复合体II;CTLA-4:细胞毒性T淋巴细胞相关抗原4;IDO:吲哚胺2,3-双加氧酶;DC:树突细胞;TIGIT:T细胞免疫球蛋白(immunoglobulin,Ig)与免疫受体酪氨酸抑制基序(immunoreceptor tyrosine-based inhibition motif,ITIM)结构域。
4.1.1. 调节性 T 细胞的分泌
调节性T细胞可分泌不同的抑制性细胞因子,如IL-10、TGF-β[43]和IL-35[44]。目前,认为IL-10和TGF-β可能参与调节性T细胞调控活性的观点仍存有争议。IL-10和TGF-β可缓解调节性T细胞缺陷造成的肠道炎症[45]。然而,IL-10和TGF-β在体外的中和并不会损害调节性T细胞的抑制作用[46]。Collison等[44]已证实,调节性T细胞和效应T细胞的共培养导致由EB病毒诱导基因3(Ebi3)和IL-12a组成的IL-35上调,并且IL-35的异位表达可以抑制调节性T细胞的功能。目前已充分证实,IL-35有利于消除炎症性肠病和牛皮癣[47]。多个小鼠人癌模型表明,中和IL-35可抑制肿瘤生长[48]。此外,调节性T细胞可通过分泌颗粒酶B(GZ-B)实现免疫抑制。颗粒酶B是一种能诱导靶细胞凋亡的丝氨酸蛋白酶[49]。与野生型小鼠相比,从颗粒酶B– /– 小鼠中得到的调节性T细胞的抑制能力较低[50]。
4.1.2. 细胞间的直接接触
调节性T细胞还可通过细胞间经TGF-β、CTLA-4、GITR以及半乳糖凝集素1的直接接触发挥作用。CTLA-4是一种由调节性T细胞和效应T细胞共同表达的共抑制分子。效应T细胞的活化需要CD28和CD80/CD86结合提供的共刺激信号。CTLA-4的亲和力高于CD28的亲和力,由此限制了CD28和CD80/CD86的结合[51−53]。GITR是一种共刺激分子,在调节性T细胞中组成性高表达。GITR敲除小鼠表现出调节性T细胞的功能受损[54]。属于β-半乳糖苷酶结合蛋白家族的半乳糖凝集素1是调控细胞增殖的一个负生长因子。分泌在调节性T细胞表面的半乳糖凝集素1与效应T细胞相互作用,破坏其细胞周期进程并由此诱导其凋亡。半乳糖凝集素1的阻断降低了人与小鼠中调节性T细胞的功效[55]。
4.1.3. 细胞外三磷酸腺苷
受损细胞产生的细胞外三磷酸腺苷(adenosine triphosphate,ATP)充当炎症反应的“天然辅助剂”。胞外酶CD39与CD73可将三磷酸腺苷降解为腺苷一磷酸(adenosine monophosphate,AMP),并产生腺苷(效应T细胞活化抑制剂)[56,57]。
4.1.4. 调节性 T 细胞与传统 T 细胞的竞争
IL-2是调节性T细胞和效应T细胞所需的一种生长因子。调节性T细胞本身不能产生IL-2,原因在于关键转录因子FOXP3的表达,FOXP3是IL-2基因的转录抑制因子。因此,调节性T细胞与传统T细胞之间的竞争是调节性T细胞抑制功效的潜在机制之一[58]。
4.1.5. 树突细胞
树突细胞(dendritic cell,DC)是负责将抗原呈递至T细胞的抗原呈递细胞。调节性T细胞可通过淋巴细胞活化基因3蛋白(lymphocyte activation gene 3,LAG3) [59]、T细胞免疫球蛋白(Ig)和免疫受体酪氨酸抑制基序(ITIM)结构域(TIGIT)[60]以及NRP1[61],与树突细胞相互作用,诱导产生免疫抑制性吲哚胺2,3-双加氧酶[62]。吲哚胺2,3-双加氧酶降解色氨酸并导致T细胞凋亡。
4.1.6. 氧化应激
肿瘤中的调节性T细胞在氧化应激下会死亡。值得注意的是凋亡调节性T细胞仍具有免疫抑制性,这给靶向调节性T细胞的抗肿瘤免疫疗法带来了挑战。这种机制可能是由于释放了大量小代谢物(如ATP)。通常,ATP对宿主有利,但是急死状态的调节性T细胞快速将ATP转化为腺苷,腺苷随后又与T细胞表面的受体结合并影响T细胞的功能[63]。
《4.2. 肿瘤中调节性 T 细胞的功能》
4.2. 肿瘤中调节性 T 细胞的功能
关于调节性T细胞的功能,人们最熟知的是其抑制功能。也就是,在受到TCR信号的刺激后,调节性T细胞拥有抑制多种细胞活化与增殖的能力,包括效应T细胞、B细胞、自然杀伤细胞(natural killer,NK)和树突细胞。此外,调节性T细胞的免疫抑制作用并不是具有抗原特异性,也不受主要组织相容性复合体的限制。众多研究表明调节性T细胞参与稳态调节和肿瘤免疫逃逸。来自小鼠与人类多项研究的有力证据表明,过继调节性T细胞转移可治疗由调节性T细胞缺陷或Foxp3突变诱发的自身免疫疾病[64−66]。因此,调节性T细胞对保护宿主免受失调而言,至关重要且不可缺少。
人们已经意识到,肿瘤中存在的大量调节性T细胞通常意味着预后不良[67]。为了找到肿瘤免疫与自身免疫的共同基础,Shimizu等[10]证实,通过抗CD25抗体消除调节性T细胞可有效缓解各种接种的同系肿瘤。然而,最新研究却对调节性T细胞与肿瘤预后之间的关系提出了挑战。研究者发现,根据Foxp3与CD45RA的表达,调节性T细胞可划分为三种类型:初始调节性T细胞(FOXP3lo CD45RA+ )、效应调节性T细胞(FOXP3hi CD45RA– )和非调节性T细胞(FOXP3lo CD45RA– )。初始调节性T细胞的抑制作用较弱;而效应调节性T细胞从初始调节性T细胞分化,在受到抗原刺激后,表现出极强的抑制活性且功能稳定。非调节性T细胞无法发挥抑制作用,但可分泌促炎性细胞因子。根据此分类,结直肠癌(colorectal cancer,CRC)可分为两类:一类主要由效应调节性T细胞浸润;另一类则包括大量非调节性T细胞。在前一类中,调节性T细胞意味着预后不良,而在后一类中,肿瘤微环境中存在更多调节性T细胞浸润的患者预后更好[68,69]。
事实上,调节性T细胞的存在并非总是不利的。慢性炎症疾病可促进结肠癌、肝癌和胰腺癌等特定癌症的进展这一观点得到了有力的证据支持。因此,靶向肿瘤微环境炎症网络的治疗方法也许可为肿瘤防治提供依据[70]。调节性T细胞是主要的抗炎因子之一。流行病学数据表明接触各种环境有机体的个体维持着一种预防癌症的保护性调节性T细胞表型,而对于生命初期很少接触环境有机体的卫生个体而言,由于调节性T细胞反馈环失调,其在生命后期患恶性肿瘤的风险更高[71]。
此外,越来越多的研究也显示了FOXP3+ 调节性T细胞的其他独特功能。其中一项功能是通过过氧化物酶体增殖物激活受体γ(peroxisome proliferator-activatedreceptor-γ,PPAR-γ)和白细胞介素-33-致瘤抑制蛋白2 (suppression of tumorigenicity 2,ST2)轴来调节脂肪相关的炎症和代谢过程[72,73],其产生的分泌性细胞因子双调蛋白(Amphiregulin,Areg)可促进非淋巴组织的修复[74,75]或刺激干细胞的分化[76]。这些研究结果提供了新的可能:肿瘤相关调节性T细胞的作用可能多种多样;重点关注这些额外功能的研究可能揭示抗肿瘤临床研究的治疗靶标。
《5. 免疫细胞与肿瘤微环境》
5. 免疫细胞与肿瘤微环境
无限增殖能力是肿瘤细胞的一种显著特性,这需要大量能量。细胞获取三磷酸腺苷有两种主要途径:一种是胞质溶胶中葡萄糖的糖酵解(Gly);而另一种是线粒体中通过三羧酸(tricarboxylic acid,TCA)循环进行的糖苷的氧化磷酸化。肿瘤细胞即使是在有氧环境下,也优先选择获取三磷酸腺苷及其他形式能量的糖酵解途径,这种现象被称为瓦氏效应[77]。通过糖酵解途径,肿瘤细胞产生大量代谢物,如乳酸和二氧化碳。这些产物不仅能抑制传统T细胞的作用,还可抑制树突细胞的成熟和活化。其原因在于抗肿瘤免疫细胞[如细胞毒性T淋巴细胞(cytolytic T lymphocyte,CTL)、效应T细胞和树突细胞]与肿瘤细胞一样,通过有氧糖酵解及谷氨酰胺分解途径获取能量和生物合成所需的原材料[78]。然而,调节性T细胞偏爱脂肪酸能量供应途径,并可以利用肿瘤代谢物获取能量[79]。因此,在肿瘤细胞高增殖的环境中在调节性T细胞富集,而T细胞和树突细胞丧失其功能。Chang等[80]发现肿瘤细胞和T细胞的糖代谢产物可抑制哺乳动物T细胞雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)信号通路的激活,并减少IFN-γ的数量,由此使T细胞在肿瘤微环境中显示低反应性。这些发现表明在肿瘤环境中,调节性T细胞与肿瘤细胞形成共生关系。
除了代谢原因,还有许多其他诱因可导致调节性T细胞在肿瘤微环境中聚集。肿瘤细胞与浸润性巨噬细胞可分泌各种趋化因子(如CC趋化因子配体22(CCL22)/CC趋化因子受体4(CCR4)[81]和CXC基序趋化因子12(CXCL12)/CXC趋化因子受体4(CXCR4)[82]),以募集调节性T细胞。此外,树突细胞与肿瘤环境中富集的一些抑制因子,包括IL-10、TGF-β和吲哚胺2,3-双加氧酶,可将效应T细胞转化为CD4+ CD25+ FOXP3+ T细胞并促进胸腺源性调节性T细胞的扩增。
《6. 肿瘤相关调节性 T 细胞的独有特征》
6. 肿瘤相关调节性 T 细胞的独有特征
增强对肿瘤浸润调节性T细胞独有特征的理解,对治疗效益而言非常重要。Plitas等[83]发现:相对于正常乳腺实质(normal breast parenchyma,NBP)和外周血,肿瘤驻留T细胞增多,FOXP3+ 调节性T细胞也表现出高度增殖表型。此外,更高级别与更强侵袭性的乳腺癌似乎含有更多抑制功能强大的活化调节性T细胞。纯化CD25hi CD4+ 调节性T细胞和CD25– CD4+ 效应T细胞的RNA测序分析表明,肿瘤驻留调节性T细胞和效应T细胞的基因表达模式和与外周血隔离的相应细胞的基因表达模式不同。进一步分析表明趋化因子(C-C基序)受体8(CCR8)为乳腺癌驻留调节性T细胞中表达量最高和最有差异的趋化因子受体。此外,高Ccr8/Foxp3 mRNA表达量比值与降低的无疾病生存率和总生存率水平之间具有显著相关性;这些结果表明CCR8可作为抗肿瘤免疫疗法的潜在靶。此后进行的靶向结肠癌中CCR8的研究证实了CCR8强效的抗肿瘤作用[84]。研究还发现,肿瘤调节性T细胞也强烈表达了此前与调节性T细胞不相关的几种基因,如黑色素瘤抗原H1(MAGEH1)基因和CD177。在平行研究中,De Simone等[85]和Zheng等[86]发现一系列由肿瘤内调节性T细胞优先上调的基因。值得注意的是,上述引用的三个研究中共发现了31种不同的基因,包括CTLA-4以及肿瘤坏死因子受体超家族的成员4(OX40)、GITR、4-1BB、TIGIT、ICOS和CD27。然而,由于部分非肿瘤浸润调节性T细胞中也表达了这些细胞表面标志物,因此针对这些受体的抗肿瘤免疫疗法的功能性结果仍不得而知。
《7. 抗肿瘤免疫疗法中的调节性 T 细胞》
7. 抗肿瘤免疫疗法中的调节性 T 细胞
《7.1. 与调节性 T 细胞相关的检查点阻断》
7.1. 与调节性 T 细胞相关的检查点阻断
CTLA-4、PD-1、PD-L1等免疫检查点抑制剂的临床应用可诱发多种癌症类型产生强烈而持久的反应。在使用易普利姆玛(细胞毒性T淋巴细胞相关抗原4的一种阻断抗体)治疗的肿瘤患者中,浸润肿瘤的调节性T细胞数量减少[87,88]。此外,CTLA-4信号对调节性T细胞和效应T细胞而言非常重要,因此使用治疗性抗CTLA-4抗体不仅能削弱调节性T细胞的免疫抑制功能,还能增强效应T细胞反应[89]。最近越来越多的研究对抗CTLA抗体的机制发起了挑战,这些研究表明靶向CTLA-4的抗体通过诱导抗体依赖性细胞介导的细胞毒性作用(antibody-dependent cellular cytotoxicity,ADCC)发挥作用,而非之前公认的通过抑制检查点发挥作用。值得注意的是,在结直肠癌等多种癌症中,免疫抑制调节性T细胞可能在限制癌症进展方面起着关键作用;因此,消耗这些癌症中的肿瘤浸润调节性T细胞是不明智的。抗PD-1抗体在阻断调节性T细胞方面的作用目前仍未有定论。据报道,在小鼠模型中,PD-1表达的调节性T细胞的抑制功能高于PD-1表达的调节性T细胞[90](图2)。
《图2》
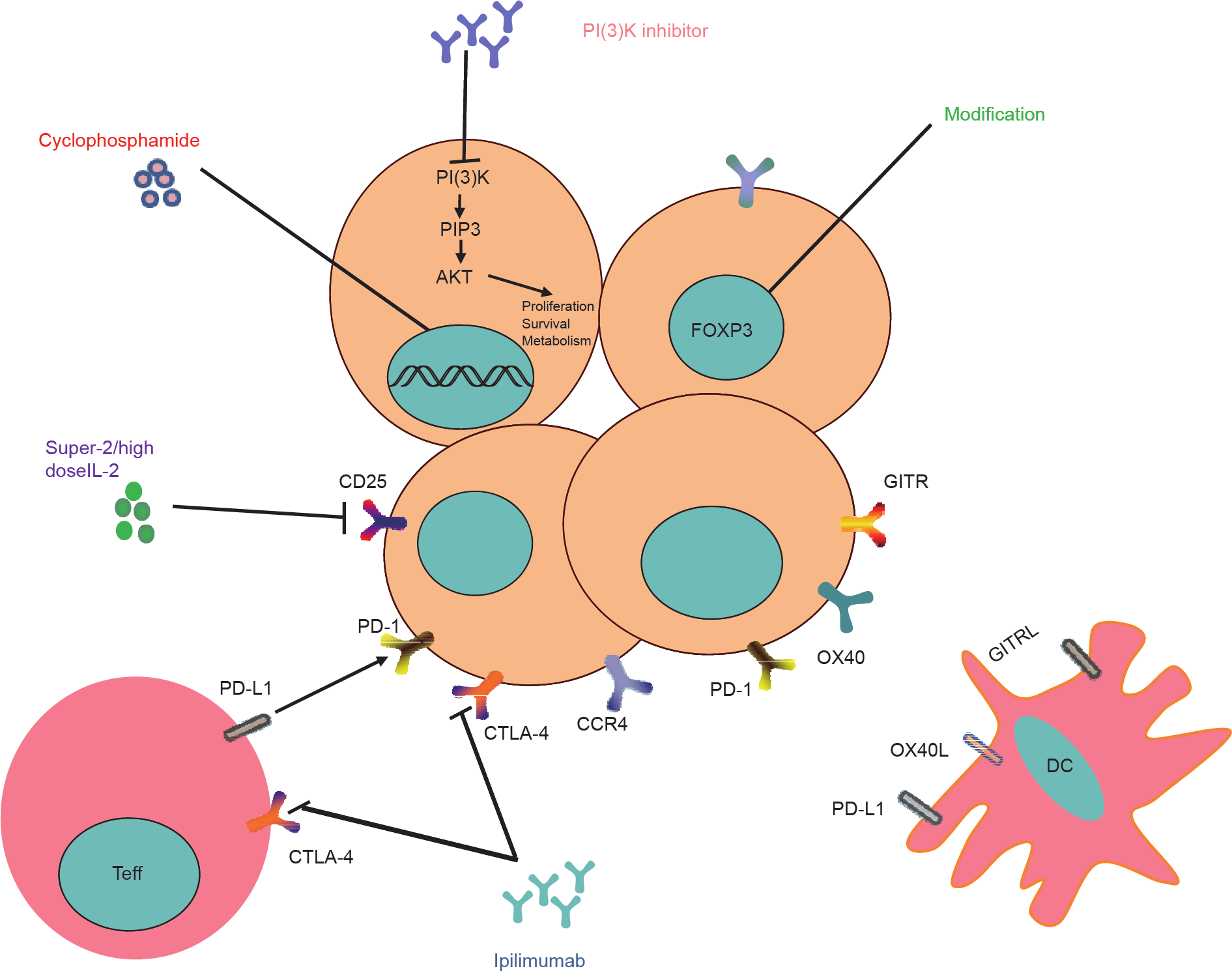
图2. 抗肿瘤免疫疗法中调节性T细胞的不同功能。PI(3)K:磷脂酰肌醇3-激酶;PIP3:3,4,5-三磷酸磷脂酰肌醇;OX40:肿瘤坏死因子受体超家族的成员4;OX40L:肿瘤坏死因子受体超家族成员4的配体;AKT:蛋白激酶B丝氨酸/苏氨酸激酶1;PD-1:程序性死亡受体1;PD-L1:程序性死亡配体1;CCR4:CC趋化因子受体4。
《7.2. 调节性 T 细胞的抑制功能》
7.2. 调节性 T 细胞的抑制功能
GITR和OX40是由调节性T细胞进行组成表达的共刺激分子。GITR和OX40的竞争性抗体,在效应T细胞活性增强后能消除调节性T细胞的抑制功能[91−94]。值得注意的是,与在肿瘤接种之前或之后立即使用抗GITR单克隆抗体相比,当肿瘤大小超过一定范围时,GITR的治疗效果更好[95]。靶向GITR和OX40的策略目前正在临床试验中。
除了表面分子,调节性T细胞特异性依赖的信号均是可用于控制调节性T细胞功能的潜在靶标。例如,据报道,磷脂酰肌醇3-激酶[phosphatidylinositol 3-kinase,PI(3)K]是一种在细胞增殖、存活与运动方面发挥核心作用的脂激酶。PI(3)K的去活化表明,其对人类白血病具有潜在的抗肿瘤活性[96]。在小鼠模型中,PI(3)K的阻断表明,其能够诱导各种实性瘤的消退[97]。
另一种策略是下调调节性T细胞关键转录因子Foxp3的表达。在小鼠中使用慢病毒Foxp3短发夹RNA,可直接导致调节性T细胞功能受损。此外,Foxp3翻译后修饰过程中涉及的分子也是好的靶标。但是,抑制Foxp3表达可能会引起不可避免的副作用。因此,需对抑制Foxp3表达进行进一步研究。
《7.3. 特异性调节性 T 细胞的消耗》
7.3. 特异性调节性 T 细胞的消耗
调节性T细胞对CD25有极高的表达,而效应T细胞在T细胞受体刺激后可短暂表达CD25。普遍公认的是,IL-2在激活调节性T细胞和效应T细胞时所扮演的双重角色限制了CD25阻断抗体与IL-2阻断抗体的临床应用。然而,众多研究者已证明抗CD25/IL-2单克隆抗体的应用潜力。Levin等[98]发现与IL-2相比,遗传修饰IL-2(super-2)与CD25 + 细胞的结合亲和力较低,诱发调节性T细胞扩增的能力也弱于细胞毒性T淋巴细胞;这一发现表明“super-2”的临床应用前景良好。不同剂量的IL-2也可能会产生迥然不同的结果。虽然采用低剂量IL-2的治疗结果不尽如人意,但对转移性黑素瘤和肾细胞癌患者使用高剂量IL-2后,部分患者出现完全肿瘤消退,而许多其他患者的无疾病间期也得到了延长,这增加了人们应用IL-2的信心[99]。抗CD25单克隆抗体的作用目前尚存争议。由于调节性T细胞和活化效应T细胞消耗的共同影响,达利珠单抗(一种靶向CD25的人源化IgG1单克隆抗体)与树突细胞疫苗接种联用治疗转移性黑素瘤,并未获得令人满意的结果[100]。有趣的是,达利珠单抗与疫苗接种联用在乳腺癌患者中产生了有利的临床反应,且无严重副作用[101]。此外还发现,在小鼠体内接种肿瘤之前使用抗CD25单克隆抗体,比在肿瘤接种之后使用产生的抗肿瘤效力更强[102]。
环磷酰胺是一种广泛用于癌症化疗的烷化剂,能够使DNA烷基化,形成DNA交联并有效地杀死增殖细胞。对拥有PROb细胞(从大鼠结肠癌得到的细胞系)的小鼠单次使用环磷酰胺后,CD4+ CD25+ T细胞的数量减少,使得延迟PROb肿瘤的生长成为可能[103]。高剂量环磷酰胺严重影响所有T细胞,而长期使用低剂量环磷酰胺会选择性地影响增殖调节性T细胞[104,105]。
由于调节性T细胞的双重功能(即肿瘤免疫逃逸与稳态保持),消除调节性T细胞后,患者往往会出现严重的自身免疫疾病。因此,如何消耗特异性调节性T细胞已经成为急需解决的一个关键问题。CCR4在调节性T细胞上有特异性表达,因此也是一个好靶标。据报道,抗CCR4抗体可选择性地消耗调节性T细胞并同时增加CD4+与CD8+ T细胞的数量[106]。而且,有力的证据表明趋化因子及其在调节性T细胞上的受体在不同类型的肿瘤中有异质性。其中包括在乳腺癌中的CCL22与CCR4,以及卵巢癌中的CCL22与CCR4、CCL28与CCR10、XCL12与CXCR4[107]。使用这些特异性表达的趋化因子可能能诱导调节性T细胞的特异性消耗。值得注意的是,消除调节性T细胞无法再次激活无反应T细胞,这表明为了重塑抗肿瘤免疫,需将调节性T细胞消耗与效应T细胞重启结合起来[108]。
《8. 结论》
8. 结论
FOXP3+ 调节性T细胞是哺乳动物免疫系统的重要组成部分,对维持免疫稳态而言至关重要。越来越多的数据表明在肿瘤微环境中,FOXP3+ 调节性T细胞是免疫系统的有效抑制因子。近年来,靶向调节性T细胞的肿瘤免疫疗法已取得有效的临床效果。然而,要使用抗肿瘤免疫疗法治愈大多数人类癌症患者仍有很长的路要走。例如,有一种策略声称可特异性消耗肿瘤中的调节性T细胞并且不会产生有害副作用,但这种策略是否有效仍有待确定。对于炎症驱动的肿瘤进展,肿瘤组织浸润调节性T细胞及其功能塑性与不稳定性仍需进一步研究。为了加深我们对肿瘤组织浸润调节性T细胞的理解,有必要开展更详细的功能研究。目前用于探索肿瘤微环境中活细胞状态的新方法(如单细胞测序)正在发展中。这些新方法的使用将极大地提高我们对于肿瘤的了解并有助于开发新的肿瘤疗法。
《Compliance with ethics guideline》
Compliance with ethics guideline
Feng Xie, Rui Liang, Dan Li, and Bin Li declare that they have no conflict of interest or financial conflicts to disclose.











 京公网安备 11010502051620号
京公网安备 11010502051620号




