

《1. 引言》
1. 引言
2019年12月,一系列不明原因的肺炎病例出现,并且由于强烈的人传人传播,病情迅速蔓延[1]。根据临床表现,肺炎被确定为病毒感染,该病毒最初被命名为2019新型冠状病毒(2019 novel coronavirus, 2019- nCoV),随后正式命名为严重急性呼吸综合征冠状病毒2(severe acute respiratory syndrome coronavirus 2, SARS-CoV-2)[2]。2020年1月30日,世界卫生组织(World Health Organization, WHO)宣布,COVID-19成为国际关注的突发公共卫生事件[3]。根据当时的数据, COVID-19的死亡率高达3.9% [4]。许多研究和报告已经确定了其平均潜伏期为4 d,最常见的4种症状有:发热、咳嗽、气短和胸闷/疼痛[5−7]。不幸的是,迄今为止还未找到有效预防和治疗COVID-19的疗法。虽然已经发现瑞德西韦和羟氯喹能够有效抑制SARS-CoV-2,但目前为止获得的数据主要都来自体外研究[8]。干扰素、洛匹那韦/利托那韦、阿比多尔、利巴韦林和血浆抗体的治疗应用也被推荐为治疗COVID-19患者的可选方案;然而,这些药物的疗效和安全性还有待进一步随机临床试验(randomized clinical trial, RCT)[9,10]的验证。
特力阿扎维林(triazavirin, TZV)是一种新型的抗病毒药物,自2015年以来一直在俄罗斯市场上销售。它是一种合成的嘌呤核苷碱基的类似物。TZV的主要作用方式是抑制病毒RNA的合成,阻止基因组片段复制 [11]。由于其多靶点的作用机制,TZV在体外和动物模型体内均具有广泛的抗RNA病毒活性:包括甲型流感病毒(H5N1等)、乙型流感病毒、蜱传脑炎和森林春季脑炎[11−13]。根据药品说明书,TZV的推荐剂量为 250 mg,每天3次,连续服用5~7 d。在TZV的II期RCT 中,患者每天口服250 mg TZV 3次,连续治疗5 d,结果发现,TZV可显著缩短流感主要临床症状的持续时间,降低流感相关并发症的发生率以及合并用药的发生率,且未见明显不良事件(adverse event, AE)报告 [14,15]。然而,TZV治疗COVID-19的疗效和安全性仍不确定。因此,我们开展了目前正在进行的多中心双盲 RCT来验证TZV治疗COVID-19的疗效和安全性。
《2. 方法》
2. 方法
《2.1. 研究设计》
2.1. 研究设计
这项临床研究是一项正在进行的多中心双盲RCT,以验证TZV相较于安慰剂治疗COVID-19的疗效和安全性。将SARS-CoV-2核酸检测为阳性的患者按1 ∶ 1的比例随机分为两组。试验组根据由中华人民共和国国家卫生健康委员会和国家中医药管理局起草的《新型冠状病毒肺炎的诊断和治疗方案(试用版7)》和《新型冠状病毒肺炎的诊断和治疗方案(试用版5)》,接受TZV加标准治疗[9,16],安慰剂组接受TZV安慰剂加标准治疗。患者接受连续7 d的治疗,并在治疗结束后的第3、7、 14天和第21天进行随访。表1概述了每次干预和随访期需完成的各项条目。本试验已在中国临床试验注册中心注册†,注册号为ChiCTR20000300001。
† http://www.chictr.org.cn/.
《表1》
表1 研究访视时间点及访视内容


RT-PCR: reverse transcription polymerase chain reaction; CT: computed tomography.
《2.2. 研究背景》
2.2. 研究背景
参加这项研究的10家医院均为三级甲等医院,具有公认的资质和资格(表2)。研究于2020年 2月14日启动,计划完成研究的日期为2020年5月31日。
《表2》
表2 研究分中心及其伦理委员会的名称

《2.3. 伦理》
2.3. 伦理
这项临床试验是根据《赫尔辛基宣言》‡的原则进行的,并遵循中华人民共和国国家卫生健康委员会的法律、法规和行政规定††。试验是在伦理委员会批准后开始的(详细资料见表2)。参与者被告知研究的风险和利益,并允许随时以任何理由停止参与研究。为了保护受试者的隐私,每个患者都通过一个独特的随机数字进行识别,除了研究人员外,患者的姓名和个人信息对其他人都保密。
‡ https://www.wma.net/what-we-do/medical-ethics/declaration-of-helsinki/.
††http://www.nhc.gov.cn/yzygj/s7659/202004/1d5d7ea301f04adba4c4e47d2e92eb96.shtml.
《2.4. 纳入标准》
2.4. 纳入标准
本试验在10个研究分中心的急诊科、隔离病房和重症监护病房(intensive care unit, ICU)进行受试者招募。研究流程如图1所示。简言之,对明确诊断为COVID-19 [经实验室实时逆转录聚合酶链反应(reverse transcription polymerase chain reaction, RT-PCR)证实为阳性]的受试者进行筛选。所有符合纳入标准且无排除标准(下面详细说明)的受试者是本研究的候选者。计划在每个研究分中心招募24名受试者,将有240名受试者参与本项研究。
《图1》
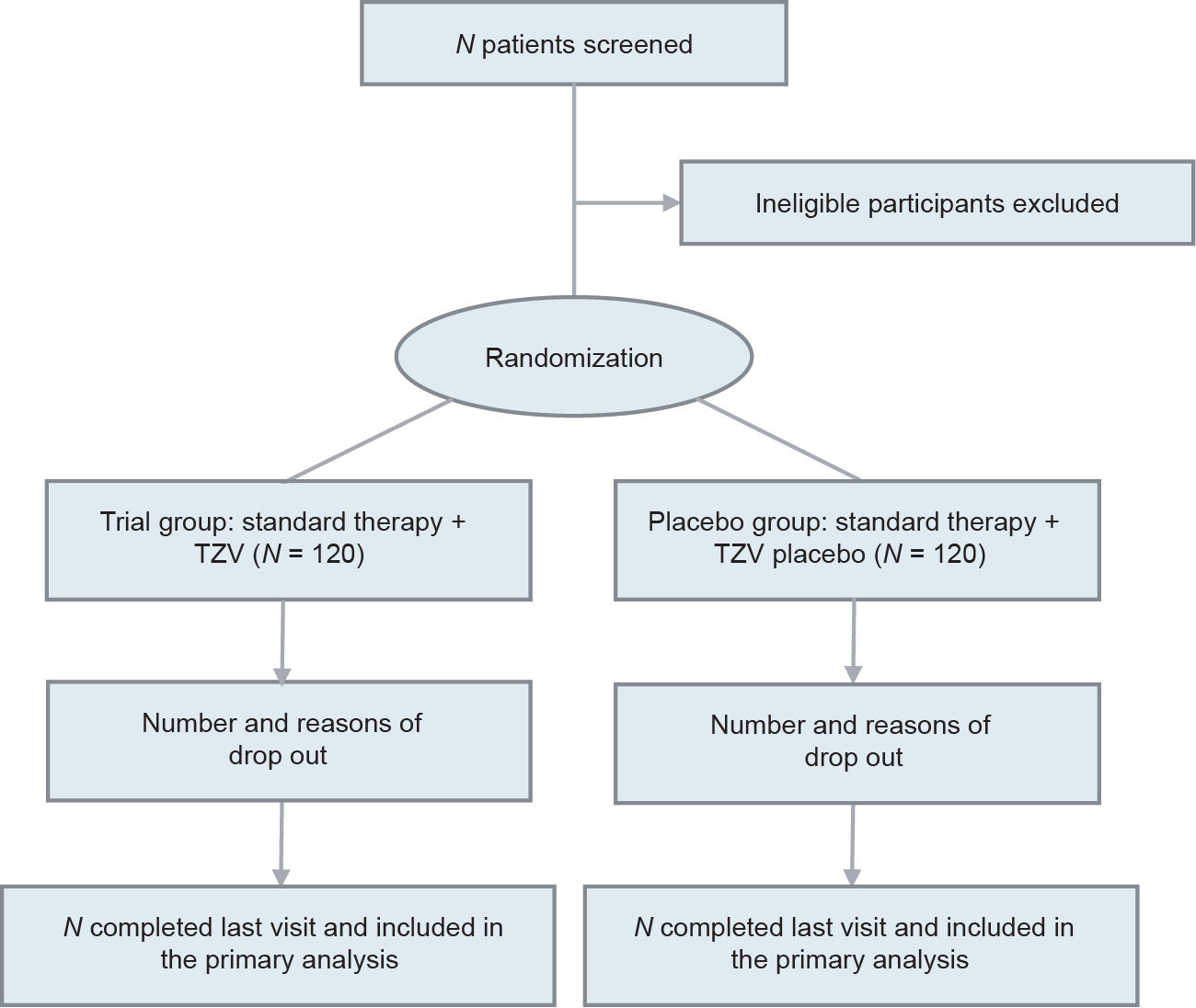
图1. 研究流程图。
纳入标准如下:①超过18岁的成年人,提供知情同意;②通过实验室实时RT-PCR证实SARS-CoV-2感染;③胸部计算机断层扫描(computed tomography, CT)成像证实肺损伤,包括肺内多发小斑片影和间质改变,以肺外带明显,或双肺多发毛玻璃影和浸润影(轻度患者可能不存在这些改变);④住院患者发热(腋温≥37.0 ℃)或有呼吸道症状;⑤症状发作与随机时间间隔≤12 d;⑥在过去3个月中没有参与其他临床研究;⑦在整个28 d的研究期间没有参与其他抗病毒研究。
排除标准如下:①由主要研究者(principle investigator, PI)判断,不适合或不能安全参与研究的患者;②根据Child-Pugh评分,评定为C级的严重肝病患者;③严重肾损害(估计肾小球滤过率≤30 mL·min−1·1.73 m−2)或持续肾脏替代治疗、血液透析或腹膜透析的患者;④严重贫血患者[血红蛋白(hemoglobin, Hgb)<60 g·L−1]; ⑤妊娠试验结果为阳性、持续妊娠或母乳喂养的妇女; ⑥有对TZV或其代谢成分过敏病史的患者;⑦未提供知 情同意的患者;⑧有可能在随机后的72 h内转移到另一 家医院的患者;⑨在筛选前30 d内参与COVID-19相关 的其他临床试验的患者。
患者可随时中止或退出试验,其退出前的数据也将包含在试验分析中。退出/脱落标准如下:①自愿退出;②干预的依从性差,患者接受治疗的时间少于3 d;③由于病情恶化而无法继续服用药物、严重不良事件(serious adverse event, SAE)的发生、并发症、致命的生理变化甚至死亡,或由研究者判断退出试验的可能性较高者;④在纳入后难以进行临床数据收集的受试者;⑤正在服用与目前研究药物有明确相互作用的药物的受试者。
试验中止标准如下:①严重偏离或违反研究方案,使药物的疗效和安全性难以评估;②在研究过程中发现严重的安全性问题;③在研究过程中没有观察到临床效果,以避免患者的延迟治疗和不必要的经济负担;④发起人建议终止研究,以保护受试者的权利和安全;⑤根据数据与安全监查委员会(Data and Safety Monitoring Board, DSMB)期中分析的结果决定终止研究。
《2.5. 干预和研究访视》
2.5. 干预和研究访视
2.5.1. 研究流程
治疗期为7 d,随访期为21 d,整个研究期共28 d。表1总结了方案中的具体访视内容和测量指标。
2.5.2. 干预措施
试验组采用标准疗法和TZV(250 mg·次−1,轻度或普通病例每日3次,共7 d;重度或危重患者每日4次,共7 d)。安慰剂组接受相同的标准治疗,并加上TZV安慰剂(250 mg·次−1,轻度或普通病例每日3次,共7 d;重度或危重患者每日4次,共7 d)。随机分组后立即给予第一剂研究药物。TZV由Medsintez(俄罗斯)制造。天晴干细胞有限公司(中国)已按照药品生产质量管理规范对TZV和安慰剂进行了重新包装并使用相同标签及外包装。TZV安慰剂胶囊仅含药用辅料,外观和气味与 TZV相同。
如果患者临床症状较轻,且胸部CT检查未发现肺炎表现,则认为该患者属于轻型患者。如果出现发热和呼吸道症状等,并且在影像学上可以看到肺炎的迹象,则认为该患者为普通型患者。重型的定义是呼吸频率≥30次·min−1,静息状态下室内空气中指氧饱和度(pulse oxygen saturation, SpO2)≤93%,动脉血氧分压(arterial partial pressure of oxygen, PaO2)/吸氧浓度≤300 mmHg (1 mmHg≈133.3 Pa),或在肺部影像学显示24~48 h内病灶明显进展>50%。危重型是指出现呼吸衰竭且需要机械通气,出现休克,或合并其他器官功能衰竭需要在重症监护病房进行监护治疗。
2.5.3. 筛查访问
筛查在招募前24 h开始,并且在筛查期间获得基线数据。体格检查和实验室检测均在当地研究中心进行,包括常规监测生命体征、指氧饱和度、心电图、动脉血气、尿常规、血常规、C反应蛋白、肝肾功能、凝血功能、降钙素原、心肌酶测定、胸部CT以及取咽拭子进行实验室RT-PCR检测。
2.5.4. 治疗访视
干预期为7 d。每日记录临床症状、生命体征、指氧饱和度、合并用药和不良事件。心电图和其他实验室检查在第3日和第7日进行。
2.5.5. 随访
对患者进行21 d的随访。如果患者住院,则每日都会记录临床症状、生命体征、指氧饱和度、合并用药和不良事件。心电图和其他实验室检查在随机第10、14、 21和28日进行。对于在随访期间出院的患者,通过电话随访记录合并用药和不良事件。
《2.6. 随机》
2.6. 随机
本研究采用多中心分层区组随机,在每个研究分中心内进行区组随机,区组长度设为4。本试验为安慰剂对照的双盲试验。符合纳入标准且不符合任何排除标准的受试者被随机分到两个治疗组中的一个:①标准治疗加TZV或②标准治疗加TZV安慰剂。在每个研究分中心,根据纳入研究时抽取的随机编号,将患者分为试验组或安慰剂组。每个研究分中心将招募24名患者,在这 10个研究分中心共计划招募240名患者。使用SAS软件版本9.4(美国SAS Institute Inc.)中的随机方法生成的随机数字表,以1∶1的比例分配受试者,试验组和安慰剂组各有120名患者。
《2.7. 电子数据收集和数据管理》
2.7. 电子数据收集和数据管理
2.7.1. 数据收集
使用电子数据采集(electronic data capture, EDC)系统管理数据。研究方案和病例报告表(case report form, CRF)相关的材料一起提供给临床数据管理中心,以便创建电子CRF,数据管理中心同时也负责后期数据统计与分析工作。对研究过程中发生的意外问题进行记录,并及时通知数据管理中心。
2.7.2. 数据管理
根据数据管理计划,数据管理员将检查在EDC系统中录入的数据,并对数据进行全面检查,以确定是否存在缺失信息、逻辑问题以及与纳入和排除标准有关的问题。数据管理员发出数据质疑,主要研究者确认后,由研究助手进行数据修改并回复质疑,数据管理员再取消此质疑。此过程将重复进行,直到数据库中的所有数据都被确认是正确的。
2.7.3. 数据监管
建立了数据与安全监查委员会来监督试验的过程和结果。DSMB由多学科的中国专家组成,包括公共卫生专家、临床专家、流行病学家、律师和社区卫生工作者。 DSMB成员定期通过微信召开会议,以审查研究方案和研究进展,并在整个研究过程中提出建议。主要目标是确保研究对象的安全和研究数据的完整性。DSMB负责就研究设计、数据质量和数据分析、研究参试者保护和研究期中分析等问题提供建议。
2.7.4. 样本量计算
鉴于有关TZV治疗COVID-19的信息有限,且临床需求紧迫,假设因死亡和其他意外情况导致的退出率低于20%,我们希望每组招募120名患者。DSMB建议进行期中分析,以监测该试验的安全性和药物的有效性,这将有助于调整样本量,并降低试验实施过程中的不确定性风险。我们采用Lan-DeMets损耗函数的O’BrienFleming界值来控制Ⅰ型错误。
2.7.5. 统计分析
统计分析将使用SAS 9.4(美国SAS Institute Inc.)进行,并且将使用意向性分析来检验两组之间的差异。分类变量将用计数和百分比进行描述,连续变量将用中位数、四分位间距或平均值以及标准差(视情况而定)来表示。在比较两组的人口统计分析中,使用Pearson χ2 检验和Fisher精确检验对分类变量进行统计。其他连续变量将使用t检验或Mann-Whitney U检验进行比较。在结局疗效分析中,采用Kaplan-Meier方法来评估不同组患者的总体生存曲线,并用log-rank检验评估生存曲线的差异。用Cox比例风险模型计算95%置信区间下的危险比。P值小于0.05将被视为具有统计学意义。对所有至少接受过一次TZV治疗的患者进行安全性评估,并比较两组的不良事件和严重不良事件的发生率。对患有糖尿病和高血压等基本疾病的COVID-19患者进行探索性亚组分析,以考虑这些基础疾病对药物治疗的有效性和安全性的影响。
《2.8. 测量结局》
2.8. 测量结局
2.8.1. 主要结局
主要结局是临床改善的时间,即从随机到相关症状恢复正常并维持至少72 h所需的天数,包括体温、呼吸频率、指氧饱和度、咳嗽缓解、肺部炎症在胸部CT图像上明显吸收。正常体温定义为腋温低于37.0 ℃。正常呼吸频率的定义是室内呼吸频率低于24次·min−1。正常指氧饱和度是指室内血氧饱和度大于94%。咳嗽缓解的定义是指重度咳嗽降至轻度或无(根据医生报告量表来评估咳嗽的重度、中度、轻度或无)。胸部CT图像上明显的炎症吸收被定义为病变吸收区域超过2/3。每个分中心胸部CT图像的医学数字成像与通信(digital imaging and communications in medicine, DICOM)数据将上传到影像中心,以确保由同一组专家进行统一评估。
2.8.2. 次要结局
(1)临床改善率:有临床改善的患者人数在所有意向性分析参试者中的比例;
(2)退热时间:退热定义为腋温低于37.0 ℃并保持 24 h或72 h以上;
(3)肺部炎症明显吸收的平均时间和人数比例;
(4)病毒核酸转阴率(连续两次阴性);
(5)第28日的病死率;
(6)重症和危重患者的转化率,这一结局将按照六分量表进行评估(表3)。受试者出院或得分下降两分被定义为从重症或危重症好转至中度、轻度或恢复。
《表3》
表3 症和危重患者的六分量表

2.8.3. 探索性结果
探索性结果包括血尿常规、凝血功能以及炎症指标(包括C反应蛋白和降钙素原)的变化。
2.8.4. 安全性结局
在整个试验过程中,将监测并记录不良事件、严重不良事件、肝功能、肾功能和合并用药。研究团队中每个人在试验开始时均接受了关于不良事件定义的培训。研究期间发生的每一次不良事件(无论是否与研究药物有关)以及随访期间的每一次不良事件都将予以报告和记录。以下不良事件相关信息将被记录:发生时间、严重程度、持续时间、采取的措施和结果。如发生严重不良事件,应在24 h内将事件报告给DSMB和当地医院的伦理委员会。
《3. 讨论》
3. 讨论
迄今为止,还没有任何药物有证据表明能够成功有效治疗由SARS-CoV-2感染引起的可危及生命的 COVID-19。目前的治疗策略基本上是经验性或尝试性的治疗和重症监护/生命支持,如检疫和隔离、早期诊断、控制感染源、减少传播的个人防护、细致的支持性护理以及对有感染患者的支持治疗[5]。目前正在中国进行一项关于瑞德西韦(一种广谱抗病毒核苷酸药物前体,以前曾用于治疗MERS-CoV和SARS-CoV感染)的 RCT [17,18],样本量大小为761人,来评估瑞德西韦治疗 COVID-19的有效性和安全性。在武汉和中国其他城市也有已经和正在进行的一系列临床试验和队列研究。在中国临床试验注册中心†或美国临床试验注册中心††已经注册了300多项有关COVID-19的干预或观察性研究,其中只有10个是RCT。本文提出的试验有望为TZV治疗COVID-19的有效性和安全性提供第一手证据。
†http://www.chictr.org.cn/
†† https://clinicaltrials.gov/
该试验的优势之一是它是一项加载试验,由国内标准疗法加上研究药物或安慰剂组成,标准治疗方案是根据国家卫生健康委员会制定的指导原则制定。这种设计既可确保方案的依从性,又最大限度地保障了参与本试验的受试者利益。另一个优势是它在10个研究分中心分别进行区组随机,在急救患者的生命支持治疗期间,既可以记录下每个分中心受试者的基本人口特征和治疗特征,又可以通过随机平衡这些特征。
我们的研究有一些局限性。首先,所有临床症状和体征的评估都是由主要研究者和研究助手在当地分中心进行的,事先没有对关于COVID-19的评估进行充分的培训。其次,没有采用统一的样本转移程序在中心实验室进行统一诊断或检测。再次,COVID-19是一种时限性疾病,目前黑龙江省有100名确诊的COVID-19患者,我们无法保证能够获得试验的预期患者数量。
《4. 结论》
4. 结论
我们设计了这项随机临床试验,以验证TZV治疗COVID-19的有效性和安全性。尽管由于样本量有限,这项研究的结果缺乏统计学意义,但结果表明, TZV可以通过减轻症状,降低合并治疗的使用而使 COVID-19患者受益。今后需要开展大样本量的研究来评估这些结果。
《致谢》
致谢
我们非常感谢参与研究的一线临床医生,他们在直接抗击疫情的同时参与了这项研究。本研究获得中国工程院COVID-19项目(2020-KYGG-01-04)的支持。
《Compliance with ethics guidelines》
Compliance with ethics guidelines
Xiaoke Wu, Kaijiang Yu, Yongchen Wang, Wanhai Xu, Hongli Ma, Yan Hou, Yue Li, Benzhi Cai, Liying Zhu, Min Zhang, Xiaoli Hu, Jingshu Gao, Yu Wang, Huichao Qin, Mingyan Zhao, Yong Zhang, Kang Li, Zhimin Du, and Baofeng Yang declare that they have no conflict of interest or financial conflicts to disclose.











 京公网安备 11010502051620号
京公网安备 11010502051620号




