

《1. 引言》
1. 引言
全球二氧化碳(CO2)排放量不断上升,这引发了国际上的广泛关注。人们也进行了一些科学研究以减少人类活动对气候变化的影响[1,2]。2016年颁布了《巴黎协定》,其重要目标是用各种方法减缓未来几十年的 CO2排放量的增长[3,4]。碳捕集与封存(CCS)技术是一种可行办法,实现这项技术的前提是掌握可行且有效的CO2捕集技术。CCS有助于各国政府完成《巴黎协定》的目标,特别是对工业行业而言,其中包括氨化肥生产、水泥窑以及钢铁制造等重要行业。如今,有许多可行的碳吸收技术,如吸收[5–7]、吸附[8–10]、膜分离[11,12] 和缩合[13,14]等,其中使用胺水溶液的化学吸收技术是商业上最为可行的碳捕集技术,它可应用于烟气中的 CO2捕集(燃烧后碳捕集)和天然气脱硫中[15,16]。
单乙醇胺(MEA)、二乙醇胺(DEA)和甲基二乙醇胺(MDEA)是大规模CO2吸收应用中常用的伯胺、仲胺和叔胺。伯胺和仲胺都能直接与CO2发生快速连续的化学反应并产生氨基甲酸酯,这大大提高了其碳吸收的动力学速率。然而,在伯胺溶液和仲胺溶液中,氨基甲酸酯的形成会导致碳吸收能力相对较低(1 mol胺吸收0.5 mol CO2),即一分子CO2与两分子的胺反应。尽管MEA和DEA的碳吸收速率相较于叔胺更快从而被广泛应用于商业中,但是,这些化合物仍会受到再生高温、腐蚀和氧化等问题的干扰[17,18]。另一方面,在叔胺溶液中,一分子CO2与一分子胺反应生成碳酸氢盐,碳酸氢盐具有很强的吸收能力(1 mol胺吸收1 mol CO2)。因此叔胺MDEA具有较高的碳吸收能力、较低的再生热和较强的抗化学氧化性[19,20]。然而,由于MDEA水溶液或叔胺溶液的碳吸收速率较慢,其不利于在商业中发展。
2007年,Vaidya和Kenig [21]合成了一种碳吸收率更高的吸收剂N,N-二乙基乙醇胺(DEEA)以代替叔胺 MDEA。之后,Chowdhury等[22,23]测试了26种叔胺的吸收性能,证明了DEEA与MDEA相比具有更高的碳吸收速率、碳吸收循环能力和更低的吸收热。尽管如此,该研究仍支持“叔胺MDEA和DEEA的碳吸收速率与伯胺或仲胺没有可比性”这一结论。
为了利用叔胺的特性,通过添加伯胺和仲胺可以解决叔胺碳吸收速率低的问题。Hafizi等[19]研究了在 MDEA溶液中添加不同的多胺以充当活性促进剂的促进效率。类似地,Gao等[6]研究了不同胺类催化剂对 DEEA水溶液的碳吸收率的影响。结果表明,胺类促进剂尽管可以有效地提高碳吸收速率,但也会大大提高再生过程中腐蚀活性和化学氧化所需的热量。近年来,有研究指出在叔胺水溶液中添加金属氧化物纳米颗粒和碳基纳米材料可以提高碳吸收能力和碳吸收速率,该方法可以避免使用胺促进剂对实验带来的不足。
Rahmatmand等[24]研究得出,仅添加0.02 wt%的碳纳米管(CNT)可使MDEA溶液的碳吸收能力提高到 23%。Komati和Suresh [25]指出,Fe3O4纳米颗粒可以降低MDEA溶液中的液-气界面的传质阻力,从而加强传质。Irani等[20]和Maleki等[26]基于氧化石墨烯(GO)和胺官能化氧化石墨烯(GO-NH2)为促进剂对MDEA 水溶液的CO2吸收能力和H2S吸收能力展开了研究。他们指出,以原始氧化石墨烯(GO)为促进剂的MDEA 溶液的碳吸收能力增强是由于其受纳米流体的物理促进机制的控制。另外,以胺官能化氧化石墨烯(GO-NH2)为促进剂的MDEA溶液不仅受益于纳米流体的物理促进机制,而且可以利用化学吸收促进的优势。Tavasoli 等[26]的实验结果表明,氧化石墨烯(GO)表面的伯胺官能团能与CO2分子发生激烈的化学反应,这可增强MDEA水溶液的碳吸收能力和碳吸收速率。
然而,由于氧化石墨烯(GO)的表面积小、孔隙率低,其活性伯胺基团的数目受到限制,从而影响其化学促进效率。本研究重点在于强调胺官能化的纳米多孔层的纳米粒子作为DEEA系统的促进剂的可能性。因此,本研究利用商业活性炭作为制备纳米多孔碳材料促进剂(NCP)的原料。商业活性炭具有表面积大、表面功能多样的优点,是一种用于制备胺基纳米流体的不可取代的材料。接着用多胺乙二胺(EDA)和聚乙烯亚胺(PEI)对合成的NCP进行后续官能化。这两种胺均具有作为促进剂必备的伯胺部分。综上,本工作探究了纳米孔碳结构可及表面积上的活性伯胺、仲胺基团,验证了它们对于DEEA水溶液的碳吸收能力和碳吸收速率提高的促进作用。
《2. 原料与方法》
2. 原料与方法
《2.1. 原料》
2.1. 原料
用作碳源的椰壳基微孔活性炭(Acticarb GC1200 GAC)和煤基介孔碳(Acticarb GS1300 GAC)购自澳大利亚Activated Carbon Technologies公司。DEEA 99.5%、4-羟基乙烷磺酸(MES, 99.5%)、4-吗啉丙磺酸(MOPS, 99.5%)、EDA和支链PEI(MW = 25 000, 99%)均购自德国Sigma-Aldrich公司。1-乙基-3-(3-二甲基氨基丙基)碳二亚胺盐酸盐(EDC, 99%)和N-羟基丁二酰亚胺(NHS, 99%)购自美国Proteochem公司。硝酸(HNO3, 70%)和硫酸(H2SO4, 98%)购自澳大利亚 Chem-Supply公司。用于CO2吸附实验的超纯氮气(N2, 99.9%)和二氧化碳(CO2, 99.9%)购自澳大利亚BOC Gas Australia公司。氧化氘(D2O, 99.9%)、乙腈(AN, 99.9%)和核磁共振(NMR)管购自Sigma-Aldrich。所有化学品均直接使用,未经进一步处理。
《2.2. 氧化纳米多孔碳材料促进剂的合成》
2.2. 氧化纳米多孔碳材料促进剂的合成
用去离子(DI)水洗涤2 g商用粒状活性炭(GAC)数次后,将其放置在60 ℃的真空烘箱中干燥过夜,再用研磨机将其完全粉碎,得到氧化NCP,然后将该活性炭细粉放入圆形烧瓶中,与100 mL浓HNO3混合。由于 HNO3与高比表面积的活性炭之间会发生氧化反应放热,注意在室温下向烧瓶中缓慢添加HNO3。另外,为了提高HNO3的氧化能力,使用磁力搅拌器强烈搅拌混合物,同时向其中缓慢加入40 mL的强氧化促进剂H2SO4。用超声破碎方法处理活性炭粉末2 h,使其更均匀地溶解在HNO3/H2SO4混合物中。然后将获得的混合物加热至 85 ℃后回流12 h,接着用1000 mL水稀释,分离出上清液,用烧结玻璃漏斗将其过滤,然后用大量沸水洗涤上清液直至pH值变为中性。分离出残余物后将其放置在60 ℃的真空烘箱中干燥过夜。标记微孔结构为NCPGC-COOH,标记介孔结构为NCP-GS-COOH。
《2.3. 氨基官能化 NCP 的合成》
2.3. 氨基官能化 NCP 的合成
为了将多胺的胺基偶联到NCP-COOH的羧基上,用 EDC-NHS交联法分两步对NCP-COOH样品进行胺官能化。首先,取1.5 g NCP-COOH加入1000 mL MES缓冲液(0.1 mol·L–1, pH 5.5)中,超声处理15 min使溶液更均匀,接着向其缓慢地加入3.3 g EDC和2 g NHS并在室温无光的条件下轻轻搅拌18 h后用烧结玻璃漏斗过滤,再用去离子水进行清洗,完全去除多孔结构上未发生反应的EDC/NHS分子。其次,将过滤后的具有胺反应性NHS酯基团(NCP-NHS)的NCP样品悬浮在500 mL MOPS缓冲液(0.1 mol·L–1和pH 7.5)中,超声处理 15 min后得到均匀溶液。接着,将500 mL EDA或PEI水溶液(10 mg·mL–1)与该溶液混合,并在室温下再搅拌 12 h,将胺反应性NHS酯基团移植到NCP的表面上。最后,多胺与NCP-NHS胺反应性NHS酯基团发生连接后,过滤反应混合物并用去离子水洗涤,去除未反应的胺,然后在60 ℃下干燥过夜,将其标记为NCP-XY,其中X 表示样品的孔隙类型(GC或GS),Y表示表面上的接枝多胺(EDA或PEI)。例如,NCP-GC-EDA代表表面接枝EDA分子的微孔碳材料促进剂。多胺在NCP表面的连接反应途径如图1所示。
《图1》

图1. NCP和多胺接枝NCP的合成原理图。
《2.4. 特性描述》
2.4. 特性描述
用红外光谱仪Bruker Tensor II(美国Bruker公司)的衰减全反射(ATR)模式收集到波长范围在 400~4000 cm–1内的信号进行傅里叶变换红外光谱(FTIR)分析,由此判定出样品中存在的化学键和官能团。用LECO TruMac CNS分析仪(美国LECO公司)测定样品的元素组成。用NETZSCH TG 209 F1 Libra分析仪(德国NETZSCH Holding公司)间接地测量官能团的数量。在温度为30~850 ℃、加热速率为10 ℃·min–1 的条件下,在氮气环境中进行热重分析(TGA)和差动热重分析(DTG)。除了在真空压力为10–7 Pa、以Al Kα为辐射源、固定光子能量为1486.6 eV的条件下,还用VG ESCALAB 220i-XL光谱仪(美国Thermo Fisher Scientific公司)分解出X射线光电子能谱(XPS)。除了在1.0 eV分辨率下对0~1400 eV范围内进行广泛扫描之外,还对C 1s谱(282~294 eV)、N 1s谱(396~408 eV)和O 1s谱(528~538 eV)进行高分辨扫描,分辨率为 0.05 eV。用雷尼绍inVia Qontor共焦拉曼显微镜系统(印度Renishaw公司),在激光激发波长532 nm、光谱分辨率1 cm–1、范围800~3200 cm–1时采集得到拉曼光谱。为了在拉曼光谱仪的工作过程中有效地固定住碳粉,实验中将样品溶于乙醇中,涂在硅片上,并在室温真空下干燥2 h。用FEI Quanta 200 ESEM FEG仪器(美国)在 20 kV和30 kV操作电压下拍摄扫描电子显微镜(SEM)图像。用装备了He-Ne离子激光器(λ= 633 nm)的马尔文纳米粒径电位分析仪(英国Malvern Panalytical公司)测量出Zeta电位和动态光散射(DLS)。借助NMR 核磁共振波谱分析法(Agilent 500 MHz;美国Agilent Technologies公司)识别出胺溶液吸收CO2过程中形成的物质。其中,13C核磁共振(13CNMR)的延迟时间为 20 s,扫描次数为128次。质子核磁共振(1H NMR)的延迟时间为2 s,扫描次数为32次。每个核磁共振(NMR)管均包含600 μL D2O(作为溶剂)、100 μL样品和10 μL 乙腈(作为内部参照)。利用Micrometrics 3Flex设备(MicroMetrics,美国)在77 K进行氮气吸附,测量出其表面积和孔体积。利用BS/IP/RF U型管逆流毛细管黏度计(ASTM D445-06)测量胺溶液和制备的纳米流体的黏度。
《2.5. DEEA-NCP 纳米流体的制备》
2.5. DEEA-NCP 纳米流体的制备
在所有实验中,DEEA水溶液的浓度保持在 3 mol·L–1不变。将350 g DEEA溶解在去离子水中,并加水稀释至1 L。取0.1 g NCP样品分散在100 mL 3 mol·L–1 DEEA水溶液中,超声处理30 min后制备出DEEA-NCP 纳米流体。标记所得的纳米流体为DEEA-X-Y,其中X 表示NCP的孔隙类型(即GC或GS),Y表示NCP的表面功能(即COOH、EDA或PEI)。
《2.6. 二氧化碳吸收能力的测量》
2.6. 二氧化碳吸收能力的测量
用室内气液平衡(VLE)设备定量监测纳米流体的吸收情况。操作步骤和测量精度的具体内容可参见我们先前的研究[5,27]。图2为CO2吸收装置的示意图。这是一个典型的碳吸收实验。取100 mL纳米流体装入不锈钢反应器(可用容积为325 mL)。然后,完全密封反应器,将其放入水浴,连接气体注入系统。在碳吸收实验开始之前先清洗系统,在70 ℃下利用纯净氮气吹扫15 min,除去溶解在DEEA-NCP纳米流体中的空气和其他干扰气体分子。接着,用纯净N2将反应器加压至 175 kPa,水浴温度调到合适温度(通常为40 ℃)。当反应器温度达到设定值且压力稳定后,用纯净CO2气体对缓冲罐(500 mL)加压,记录最终压力(P1)。然后,将加压的CO2从缓冲罐注入反应器,再次记录缓冲罐的新压力(P2)。
分别在CO2缓冲罐和反应器上安装高精度(全量程精度为0.08%)的微型硅压力传感器:Omega, PX409- 150 GUSBH压力传感器(压力范围为0~1030 kPa)和Omega, PX409-100 GUSBH传感器(压力范围为 0~690 kPa),以便动态监测和记录设备的压力变化情况。在水浴容器上安装数显浸入式加热器循环器Ratek, TH7000(Ratek Instruments,澳大利亚)和精度为±0.1 ℃的热电偶Omega,TJ-USB-K1(英国OMEGA Engineering公司),以便高精度控制水浴的温度和监测反应堆温度。
注入VLE反应器的CO2总摩尔数的计算式为:
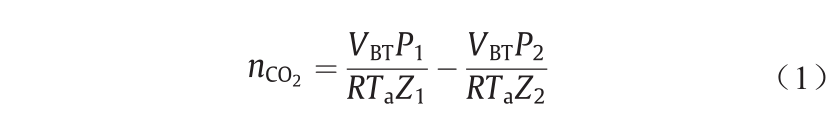
式中,P1代表注入CO2之前缓冲罐的压力;P2代表CO2注入缓冲罐后的压力;VBT表示缓冲槽的可用容积;Ta代表环境温度;R代表摩尔气体常量;Z1、Z2分别对应于P1、P2时CO2的压缩因子。
不同CO2分压下的压缩系数的计算式为SoaveRedlich-Kwong (SRK)状态方程[公式(2)][1]:

式中,P代表压强;T代表温度;Z代表压缩系数。所有必要系数都可以通过方程式(3)~(5)求得:
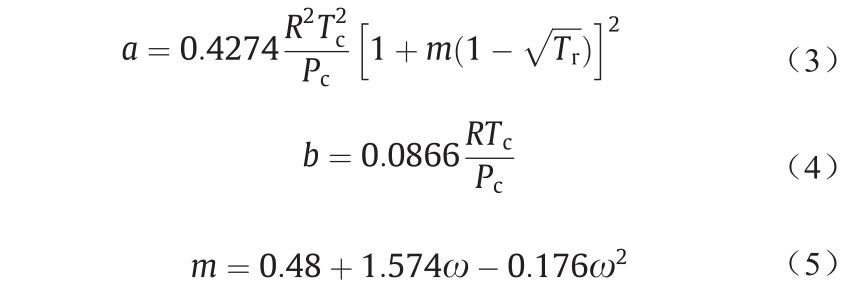
式中,Tc代表临界温度;Tr代表对比温度;Pc代表临界压力;ω代表离散系数。
VLE反应器内CO2分压( )的计算式为:
)的计算式为:
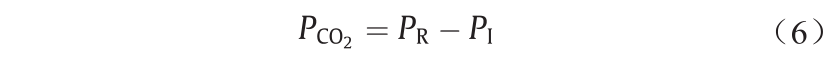
式中,PR代表反应器压力;P1代表注入VLE反应器的初始N2 压力。
因此,气体相[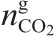 ,式(7)]和液体相[
,式(7)]和液体相[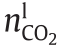 ,式(7)] 中CO2的总摩尔数由以下方法得到:
,式(7)] 中CO2的总摩尔数由以下方法得到:

式中,Vg代表反应器内气体体积;TR代表调整后的反应器温度;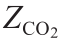 代表
代表 对应的压缩因子。
对应的压缩因子。
在每个CO2注入循环内,吸收到进入DEEA-NCP纳米流体中的CO2总量可通过公式(9)和公式(10)计算:

式中,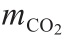 为CO2的质量摩尔浓度(每千克胺溶液中的CO2摩尔数);
为CO2的质量摩尔浓度(每千克胺溶液中的CO2摩尔数);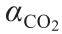 为CO2负荷量(每摩尔胺产生的CO2的物质的量); wsol为溶液的质量(kg);namine为溶液中胺的总物质的量。
为CO2负荷量(每摩尔胺产生的CO2的物质的量); wsol为溶液的质量(kg);namine为溶液中胺的总物质的量。
为了测定加入纳米多孔碳材料促进剂后对CO2的吸收速率及其提高程度,分别采用公式(11)和公式(12)计算。

式中,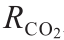 为CO2的吸收率;P为操作压力(kPa);t为时间(min);ΔP(如50 kPa、100 kPa、150 kPa、200 kPa、250 kPa、 300 kPa)为初始气体通过吸收实验后的压降(kPa)。
为CO2的吸收率;P为操作压力(kPa);t为时间(min);ΔP(如50 kPa、100 kPa、150 kPa、200 kPa、250 kPa、 300 kPa)为初始气体通过吸收实验后的压降(kPa)。
在循环的吸附-解吸CO2实验中,将CO2饱和的 DEEA-NCP纳米流体倒入圆形烧瓶中,在回流条件下加热3 h。然后,将再生的DEEA-NCP纳米流体自然冷却至室温,再次用于CO2吸收实验。再生效率定义如下:
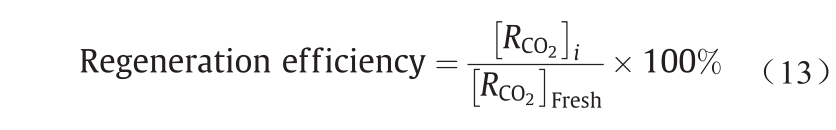
式中,i 为每个循环的编号。
《图2》

图2. 测量二氧化碳吸收量的气液平衡(VLE)装置示意图。
《3. 结果与讨论》
3. 结果与讨论
《3.1. NCP 的特性》
3.1. NCP 的特性
图3(a)和(b)显示了本工作中商用活性炭和所有合成NCP的FT-IR光谱。简而言之,以1750 cm–1和 1150 cm–1为中心的吸收带分别与C=O键和C–O键的振动有关[28,29]。此外,在1570 cm–1和1380 cm–1附近的吸光度对应于N–O和C–N伸缩振动[29,30]。与商用活性炭(即AC-GC和AC-GS)相比,NCP-GC-COOH和 NCP-GS-COOH对所有四种C=O、C–O、N–O和C–N振动均具有更强的吸收带。具体来说,C=O/C–O和N–O/ C–N特征峰的强化表明羧基(–COOH)和亚氮(–NO2)官能团成功植入纳米孔碳表面,这是HNO3氧化/硝化的直接结果[30,31]。使用EDC-NHS交联剂功能化EDA和 PEI后,与羰基键(C=O)相关的吸收带强度显著降低。同样,在聚胺与先前注入的羧基结合后,位于1150 cm–1 处的C–O振动带的强度降低。这些观察结果证实了使用 EDC-NHS交联剂能成功地将羧基转化为酰胺基,从而将多胺固定在纳米孔碳表面[32–35]。
为了获得合成NCP表面附着的官能团的其他信息,我们结合了TGA/DTG与元素分析进行报道。根据商用活性炭和功能化NCP的TGA(见附录A中的图S1)和 DTG(见附录A中的图S2)曲线,重量损失可以分为三个不同的区域。在90~110 ℃左右第一个重量损失区是由水分蒸发引起的[10]。含氧官能团(即–COOH和– OH)的分解开始于180 ℃,并持续到更高的温度。除了在300~330 ℃分解的氧基外,在EDA和PEI接枝的 NCP样品中还观察到另一个峰值(见图S1和图S2),持续到600 ℃,这主要是由胺基分解所导致的[30]。因此,180~600 ℃之间的重量损失被认为是得出官能团数量的一个指标。结果如图3(c)和(d)所示。初始的AC-GC和AC-GS分别减少了4.9%和2.1%的重量,这可以归因于它们固有的羧基(–COOH)和羰基(– OH)官能团的存在。HNO3氧化的NC-GS-COOH的减重率(20.0 wt%)显著高于NCP-GC-COOH的减重率(12.7 wt%)。有趣的是,接枝多胺后,微孔NCP-GCEDA的减重(18.1 wt%)大于NCP-GC-PEI(17.4 wt%),而聚胺功能化的介孔NCP-GS则表现出相反的行为。介孔EDA接枝NCP-GS-EDA的减重率(20.2 wt%)比PEI 接枝NCP-GS-PEI的减重率(25.8 wt%)低27.7%。由于 PEI相对分子质量大、旋回半径大,它实际上无法进入 NCP-GC结构的微孔,而EDA分子则成功地负载了高接枝转化率的微孔。同样地,不同的NCP样品元素分析的结果(表1)证明商业AC-GS碳表面存在固有的氧官能团、高浓度硝酸氧化后其表面氧含量显著增加(即 NCP-GC-COOH和NCP-GS-COOH)、聚胺结合后其表面含氮量显著增加(即NCP-GC-EDA、NCP-GC-PEI、 NCP-GS-EDA和NCP-GS-PEI),这些与先前做出的解释相符合。
《图3》
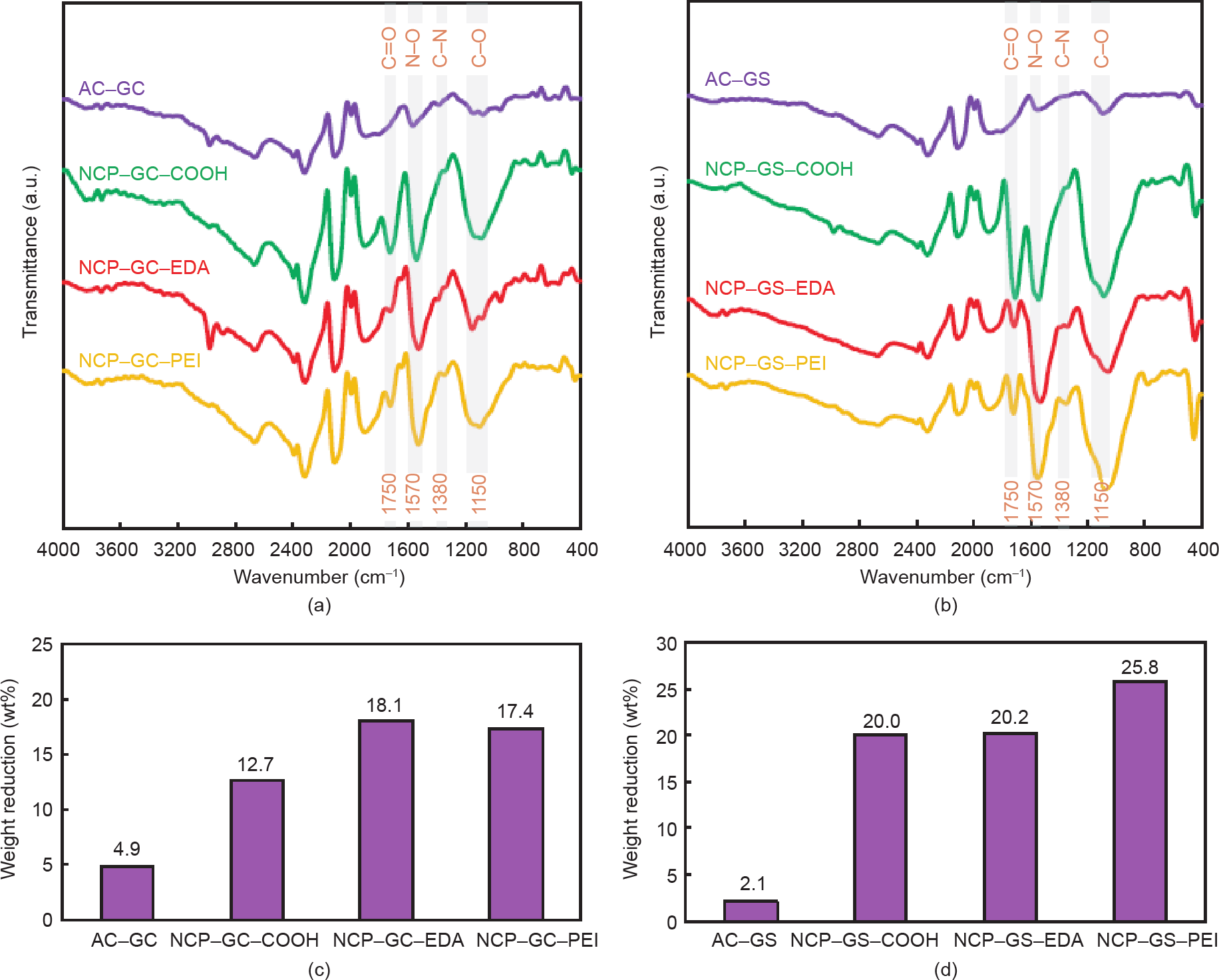
图3. (a)、(b)FT-IR光谱;(c)、(d)180~600 ℃在N2气氛下合成NCP的TGA减重率。
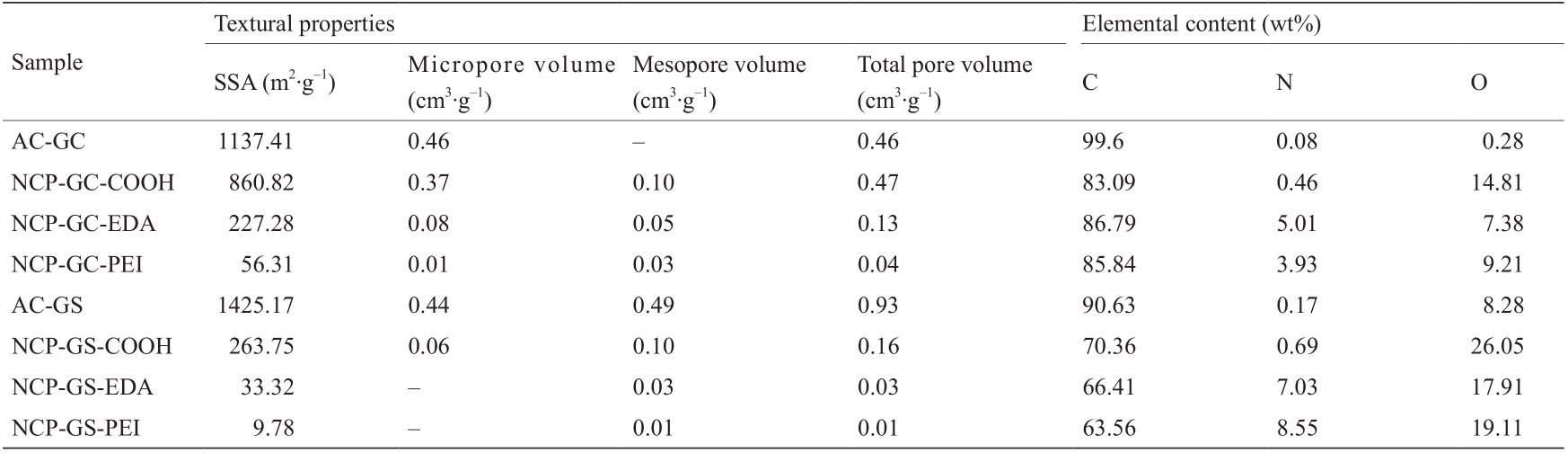
表1给出了AC(即AC-GC和AC-GS)和制备的NCP 促进剂的质构特性。可以看到,煤基AC-GS样本比椰基 AC-GC(SSA为1137.41 m2 ·g–1,孔隙体积为0.46 cm3 ·g–1)具有更大的比表面积(1425.17 m2 ·g–1)和孔隙体积(0.93 cm3 ·g–1),但是在NCP-GS-COOH中,其比表面积和孔隙体积仍受到表面氧化的显著影响,两个数值分别降至263.75 m2 ·g–1和0.16 cm3 ·g–1。相比之下,NCP-GCCOOH促进剂具有高比表面积(860.82 m2 ·g–1)和孔隙体积(0.47 cm3 ·g–1),使其成为较好的胺接枝孔基。因此,在4个多胺接枝促进剂中,NCP-GC-EDA(227.28 m2 ·g–1 和0.13 cm3 ·g–1)的质构特性最佳,NCPGS-PEI (9.78 m2 ·g–1 和0.01 cm3 ·g–1)的质构特性最差。
《表1》
表1 制备不同AC和NCP样品的质构特性和元素分析
为了进一步探究AC和NCP样品的结构,我们使用了拉曼光谱进行分析。如图4(a)和(b)所示,所有样品均在1350 cm–1(D波段)和1590 cm–1(G波段)附近出现两个峰,为碳结构的特征指标。D波段表现为 sp3 无序/非晶态模式,G波段对应于碳结构的sp2 键拉伸 [29,30,36]。因此,D波段与G波段的强度比(ID/IG)可以表明碳基纳米材料的结构紊乱度(见附录A的表S1)。 AC-GC和AC-GS均显示出较高的ID/IG值,它们分别为 1.12和1.14,这表明商用微孔和介孔活性炭的结构存在高度缺陷。NCP-GC-COOH(1.04)和NCP-GS-COOH (0.96)样品经HNO3表面氧化后,ID/IG值下降,这是因为在严苛的氧化条件下多孔碳结构的部分出现了石墨化[37]。然而,NCP-GC-COOH和NCP-GS-COOH降低的ID/IG值与之前的文献内容数据之间仍具有可比性 [8,28–30]。另一方面,当多胺通过纳米多孔碳的结构固定时,ID/IG值从NCP-GC-COOH (1.04)增加到NCP-GCEDA (1.06)和NCP-GC-PEI (1.07)。相似的趋势是,多胺功能化的NCP-GS-EDA (1.05)和NCP-GS-PEI (0.98)的ID/IG值均高于NCP-GS-COOH (0.96),这表明表面接枝聚胺过程出现了新的缺陷。
除FT-IR、TGA、元素分析外,XPS也可作为表面定量分析技术,以测量NCP表面上不同官能团的数量。图4(c)为XPS的测量光谱图以及NCP-GC-COOH和 NCP-GC-EDA表面氧/氮含量。由XPS测量光谱图4(c)所示在285 eV、397 eV和532 eV附近有三个主要峰值,分别代表C 1s、N 1s和O 1s [38]。根据表面总原子分析图4(d)显示,NCP-GC-COOH的氧含量从14.55 at%下降到12.46 at%,而NCP-GC-EDA的氮含量从1.83 at%增加到6.44 at%;这些结果初步证实了胺以酰胺形式缔合在表面上。为了详细研究胺的功能化机制,我们记录了不同部分的高分辨率光谱图,见图4(e)~(h)和附录 A中的图S3。根据图4(e),NCP-GC-COOH样品的光谱中显示了两个峰,分别是C–O–H键(含量8.22 at%;峰位置533.2 eV)和C=O键(含量5.88 at%;峰位置 531.6 eV),二者均为羧基的主要成分。另外,在NCPGC-COOH的高分辨率N 1s光谱图4(g)中,–NO2(含量为0.77 at%;峰位置在405.6 eV)和C–N(含量为 1.06 at%;峰位置在399.3 eV)峰均存在,这表明在表面氧化过程中HNO3将碳结构硝化了[39]。同样,NCPGC-EDA的高分辨率O 1s图谱[图4(f)]显示,C–O–H含量下降到4.97 at%,相反,C=O含量上升到7.09 at%。此外,如图4(h)所示,NCP-GC-EDA的高分辨率N 1s 图谱中出现了两个新的酰胺/–NH2峰(含量为5.28 at%;峰位置在399.8 eV)和–NH2峰(含量为0.8 at%;峰位置在402.2 eV)。这些变化表明,当多胺交联在NC-GCEDA的羧基上后,多胺在其表面形成了酰胺基。
《图4》

图4. (a)微孔样品的拉曼光谱图;(b)介孔样品的拉曼光谱图;(c)全XPS测量;(d)高分辨率下定量元素分析;(e)、(f)NCP-GC-COOH和 NCP-GS-EDA纳米促进剂的O 1s谱;(g)、(h)NCP-GC-COOH和NCP-GS-EDA纳米促进剂的N 1s谱。
图5(a)~(p)显示了商用活性炭、所有氧化和胺功能化处理前后NCP的SEM图像。图像显示,经过 HNO3/H2SO4处理后,碳的颗粒直径略有减小,这表明了剧烈氧化后碳结构会被分解成更小的单元。然而,在不同官能团结合前后,所有样品(无论是GC结构还是 GS结构)的形态几乎都是相同的。因此,硝酸氧化不影响NCP的结构形态,并且胺结合后没有观察到聚合现象。
《图5》

图5. (a)、(b)微孔AC-GC的SEM图像;(c)、(d)介孔AC-GS碳的SEM图像;(e)、(f)NCP-GC-COOH的SEM图像;(g)、(h)NCP-GS-COOH 的SEM图像;(i)、(j)NCP-GC-EDA的SEM图像;(k)、(l)NCP-GS-EDA的SEM图像;(m)、(n)NCP-GC-PEI的SEM图像;(o)、(p)NCP-GSPEI启动子的SEM图像。
附录A中的图S4为通过DLS测量得到的不同NCP的平均粒径分布。结果清楚地表明实验所制备的NCP样品的平均直径均小于400 nm,远小于SEM图中所显示的数值(1~2 μm)。这种差异是由于DLS和SEM的操作方式的不同产生的。在SEM显微镜中,扫描方式是在固态下进行的,这可以显著加速颗粒的团聚速度并增加团簇的尺寸。另外,在DLS技术中,颗粒在开始测量前通过超声波在溶液中充分分散,这使得DLS成为确定类似体系平均粒径的更好的表征方法。
《3.2. NCP 样品的稳定性》
3.2. NCP 样品的稳定性
为了评估NCP的纳米流体最关键的特征因素之一——稳定性,我们进行了ζ 电势研究。图6(a)~(h)显示了所有制备的具有不同功能的DEEA-NCP纳米流体在纳米多孔促进剂表面的ζ 电位值以及它们在不同时间间隔的稳定性照片。通常,ζ 电位表示悬浮在液相中的固体颗粒/团簇的静电荷(吸引电荷或排斥电荷)。因此,ζ 电位值可以间接代表纳米流体中悬浮的纳米材料的稳定性。特别是,极高的(大于10 mV)和极低的(小于–10 mV)ζ 电位值都可以表明悬浮的纳米材料在溶液中是完全稳定的。相反地,落在极限值之间的值通常显示出纳米材料的不稳定性和聚结/沉淀的趋势。如图6(a)和(b)所示,微孔NCP-GC-COOH和介孔NCP-GS-COOH表现出较高的负ζ 电位值,分别为–11.98 mV和–33.42 mV。这是因为其中存在酸性官能团–COOH和–OH。此外,NCP-GC-COOH的ζ 电位值比NCP-GS-COOH样品的ζ 电位值高约两倍,这表明椰子作为碳源比煤作为碳源更适合用于表面功能化和制备NCP。正如预期的那样,NCP-GC-COOH和NCP-GSCOOH促进剂可以在五天内保持悬浮状态的稳定。通过酰胺形成反应将多胺连接在羧基上后,去质子化胺基的正电荷使得ζ 电位值增加。ζ 电位值为–8.26 mV的NCP-GC-PEI和ζ 电位值为–5.62 mV的NCP-GS-EDA 表面上多胺基团的强正电荷使粒子的ζ 电位值提高到–10 mV以上,从而导致悬浮液不稳定,结果如图6(g)和(h)所示。与之相反的是,由于表面上的官能团分布最优,NCP-GC-EDA和NCP-GS-PEI促进剂在溶液中可以稳定保持5 d,它们的ζ 电位值分别为–26.08 mV 和–10.02 mV。
《图6》
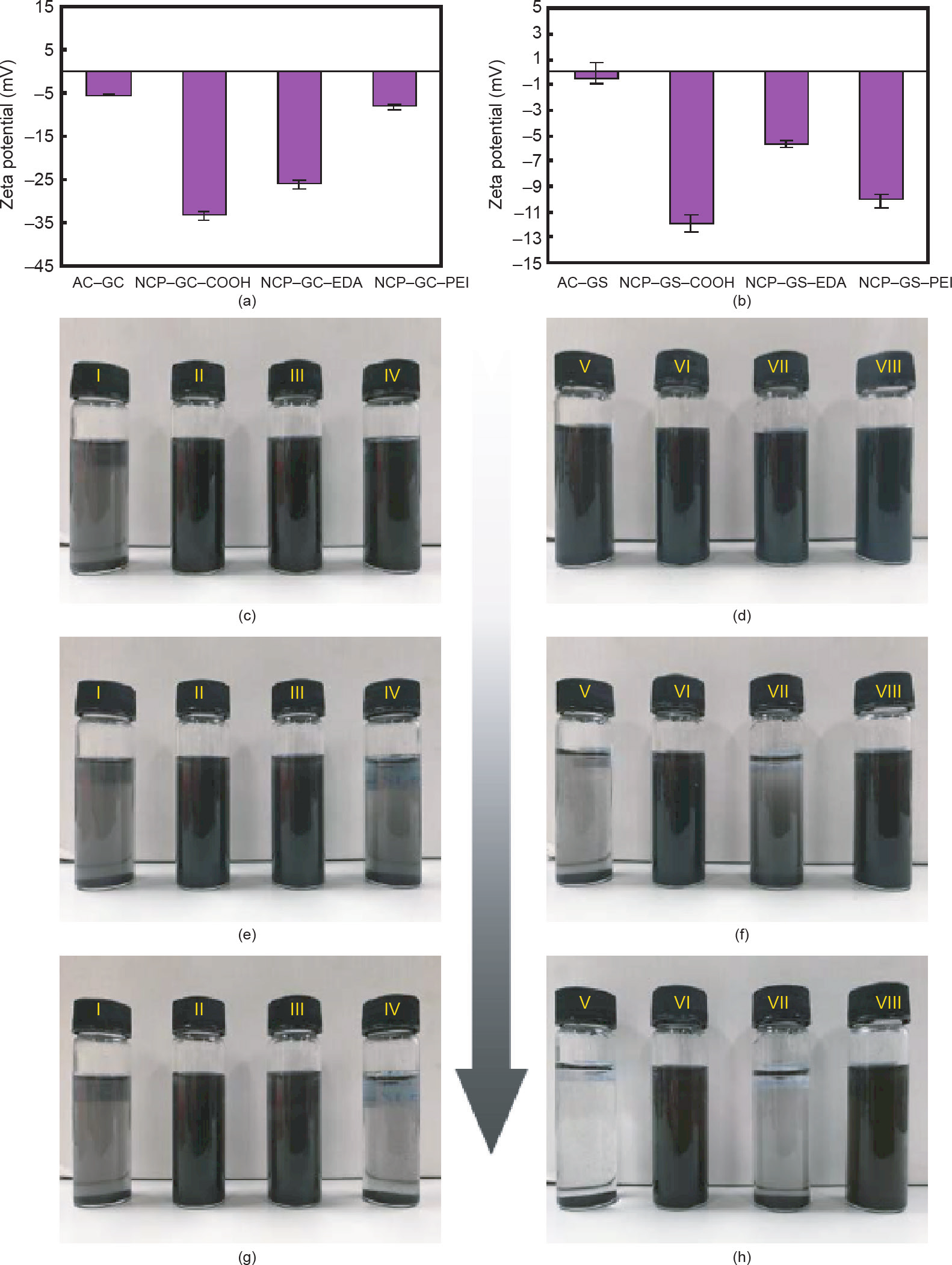
图6. 制备的含有不同成分的DEEA纳米流体的ζ 电位分析。(a)GC;(b)GS碳基促进剂;AC-GC(I)、NCP-GC-COOH(II)、NCP-GC-EDA(III)、 NCP-GC-PEI(IV)、AC-GS(V)、NCP-GS-COOH(VI)、NCP-GS-EDA(VII)和NCP-GS-PEI(VIII)促进剂,分别经声波处理1 h (c,d)、1 d (e,f)和5 d (g,h)。
《3.3. DEEA-NCP 纳米流体的 CO2 吸收动力学》
3.3. DEEA-NCP 纳米流体的 CO2 吸收动力学
作为一种叔胺,DEEA无法直接与CO2分子反应生成氨基甲酸酯。叔胺与CO2的反应受间接水解反应控制,生成碳酸氢盐和质子化胺。尽管叔胺具有相当大的优势(如高吸收能力和低解吸热),但大多数叔胺具有低CO2 吸收率,这仍阻碍了它们的普遍使用。在这项研究中,为了量化不同NCP对在DEEA水溶液中吸收CO2的速率的促进作用,在附录A中的表S2和图7(a)、(b)中表明了不同DEEA-NCP纳米流体的CO2压降随时间变化的关系。由于PEI分子包含许多伯胺基和仲胺基,因此,与仅包含一个伯胺基和仲胺基的含有EDA接枝的纳米多孔促进剂相比,NCP-GC-PEI和NCP-GS-PEI的CO2吸收率应该更高。尽管预期如此,但在相似条件下,含有PEI接枝NCP的DEEA-NCP纳米流体的CO2速率增强效果却比带有EDA接枝NCP的DEEA-NCP纳米流体差。根据图7(b),DEEA-GS-PEI纳米流体的CO2吸收曲线在整个吸收时间内与常规的DEEA溶液的曲线有相当大的重叠,这表明了NCP-GS-PEI作为促进剂的性能较差。 PEI接枝的DEEA-GS-PEI纳米流体的低效率表明,PEI 分子会阻塞多孔结构。因此,在DEEA-GS-PEI纳米流体中,多胺的大部分伯胺和仲胺位点几乎都无法被接触到,从而导致增强效果不明显。
《图7》
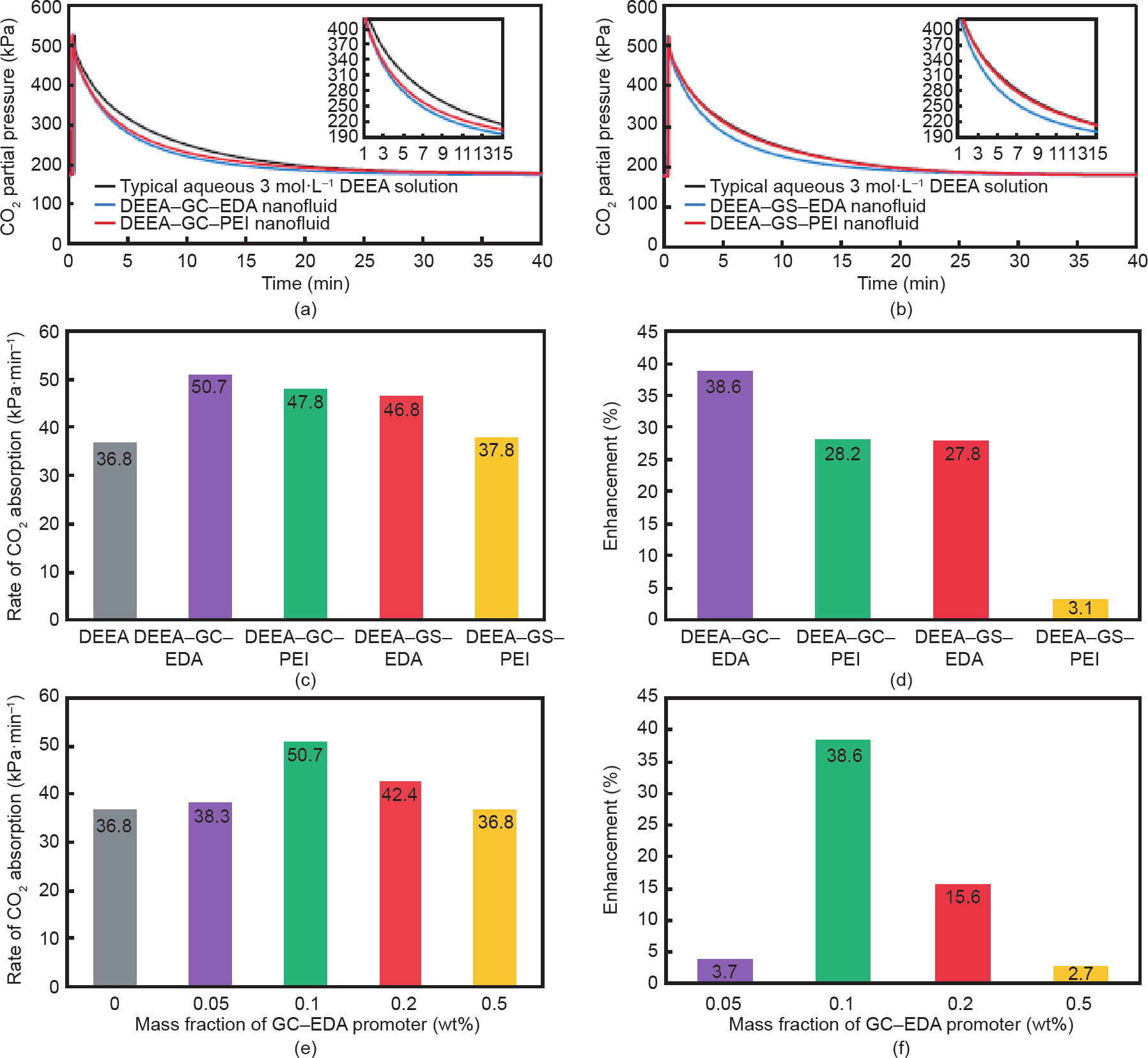
图7. 具有DEEA-GC-EDA和DEEA-GC-PEI(a)及DEEA-GS-EDA和DEEA-GS-PEI(b)纳米流体的典型3 mol·L–1 DEEA水溶液在40 ℃和0.1 wt% 纳米促进剂下的VLE实验数据;DEEA水溶液和DEEA-NCP纳米流体的CO2吸收率(c)和增强比(d);GC-EDA促进剂含量对DEEA-GC-EDA 纳米流体在40 ℃时CO2的吸收速率(e)和增强比(f)的影响。
相反,如图7(c)和(d)以及附录A中图S5和S6所示,与含有介孔促进剂的纳米流体相比,含有微孔催化剂的纳米流体表现出更优异的性能。在所有制备的DEEANCP纳米流体中,DEEA-GC-EDA纳米流体表现出最佳性能,并成功地将CO2吸收率提高到50.7 kPa·min–1,比商用DEEA溶液(36.8 kPa·min–1)高38.6%,其次是DEEA-GC-PEI(47.8 kPa·min–1)、DEEA-GS-EDA (46.6 kPa·min–1)和DEEA-GS-PEI(37.8 kPa·min–1),它们的CO2吸收率分别比商用DEEA溶液提高了 28.2%、 27.8%和3.1%。因此,在具有不同特性的合成NCP中,具有EDA接枝微孔结构的NCP-GC-EDA促进剂具有最佳特性。因此,使用NCP-GC-EDA作为叔胺溶液的促进剂,不仅可以减少叔胺溶液在吸收柱中达到恒定的吸收能力所需的停留时间,而且还可以降低溶剂循环速率,进而降低热再生所需的能量。
《3.4. NCP-GC-EDA 浓度的影响》
3.4. NCP-GC-EDA 浓度的影响
由于NCP-GC-EDA在所有具有不同多胺功能的微孔和介孔的NCP中均表现出最佳的CO2吸收速率增强性能,因此选择了纳米孔NCP-GC-EDA促进剂来评估碳质促进剂浓度对CO2吸收速率的促进作用。我们分析了具有四种不同质量分数的NCP-GC-EDA促进剂(0.05 wt%、 0.1 wt%、0.2 wt%和0.5 wt%)的DEEA-GC-EDA纳米流体对CO2吸收浓度的促进作用。CO2吸收速率和增强比都已在图7(e)和(f)以及参阅附录A中的图S7(a)中给出。如图所示,在含有0.05 wt%促进剂的DEEAGC-EDA纳米流体中,CO2吸收速率从36.8 kPa·min–1提高到38.3 kPa·min–1,提高了3.7%。但是,将NCP-GCEDA促进剂的浓度提高到0.1 wt%后,纳米流体中CO2 的吸收速率显著提高,高达50.7 kPa·min–1,这比不使用任何促进剂的DEEA水溶液要高38.6%。有趣的是,具有0.2 wt%(CO2吸收速率为42.4 kPa·min–1)和0.5 wt% (36.8 kPa·min–1)促进剂的DEEA-GC-EDA纳米流体的 CO2吸收速率远低于具有0.1 wt%(50.7 kPa·min–1)促进剂的DEEA-GC-EDA纳米流体的CO2吸收速率。NCPGC-EDA促进剂对CO2吸收速率的不利影响可能与高浓度纳米材料下纳米流体的高黏度有关(另请参见附录A 中的图S8)。这一解释与Dharmalingam等[40]的研究结果一致,他们报道了纳米颗粒对纳米流体的黏度及其相应的CO2吸收性能的巨大影响。Dharmalingam等认为,高浓度的纳米材料在水溶液中可以大大提高纳米流体的黏度,降低气液界面的传质系数,抑制了促进剂对CO2 吸收速率的积极作用。综上所述,本部分研究明确表明, 0.1 wt%的纳米孔NCP-GC-EDA促进剂作为该促进剂的最佳浓度,可以显著提高DEEA水溶液的CO2吸收速率,应避免使用更高浓度的GC-EDA。
《3.5. DEEA-GC-EDA 纳米流体中 CO2 的吸收等温线》
3.5. DEEA-GC-EDA 纳米流体中 CO2 的吸收等温线
图8以及附录A中的表S3和表S4显示了3 mol∙L–1 DEEA水溶液和DEEA-GC-EDA纳米流体(3 mol∙L–1,促进剂浓度为0.1 wt%)在不同的CO2分压和40 ℃下的CO2吸收能力。可以看出,两个液体吸收体系中的CO2 吸收趋势基本相同,并且可以通过增加CO2分压来提高 CO2的溶解度。与常用的3 mol∙L–1 DEEA水溶液相比, DEEA-GC-EDA纳米流体在CO2分压(0.1~90 kPa)的范围内明显表现出更高的CO2吸收能力,尤其是在低 CO2分压下。如图8(b)所示,在15 kPa的CO2分压下进行的简单比较,在0.1 wt%的NCP-GC-EDA促进剂的作用下,CO2吸收能力提高了13.1%,从DEEA水溶液中的0.69 mol CO2增加到DEEA-GC-EDA纳米流体中的0.78 mol CO2。换句话说,DEEA-GC-EDA纳米流体的CO2吸收能力比商用DEEA水溶液对烟道气流(在 15 kPa和40 ℃下)的CO2的吸收能力高13.1%。NCPGC-EDA促进剂对CO2吸收能力的促进效果是基于以下两个因素。首先,NCP-GC-EDA的多孔结构具有较高的表面积和孔体积,可以物理吸附溶剂中大量溶解的CO2 分子、含CO2的物质和质子化胺,从而促进碳酸氢盐的合成反应,并增加整个系统的CO2吸收能力。其次,通过NCP-GC-EDA的多孔结构,伯胺/仲胺基团可以通过氨基甲酸酯形成与CO2分子进行有效反应,促进CO2分子扩散到纳米孔结构中,并最终提高溶液的CO2吸收能力。尽管DEEA-GC-EDA纳米流体保持了其相对于商用DEEA水溶液的CO2吸收优势,但CO2吸收增强比逐渐从15 kPa的13.1%分别降至30 kPa和60 kPa的9.1%和 4.8%。NCP-GC-EDA催化剂在低CO2分压下表现更好,这可能与GC结构对物理吸附的影响以及微孔内伯/仲胺活性位点的化学促进反应有关。
《图8》
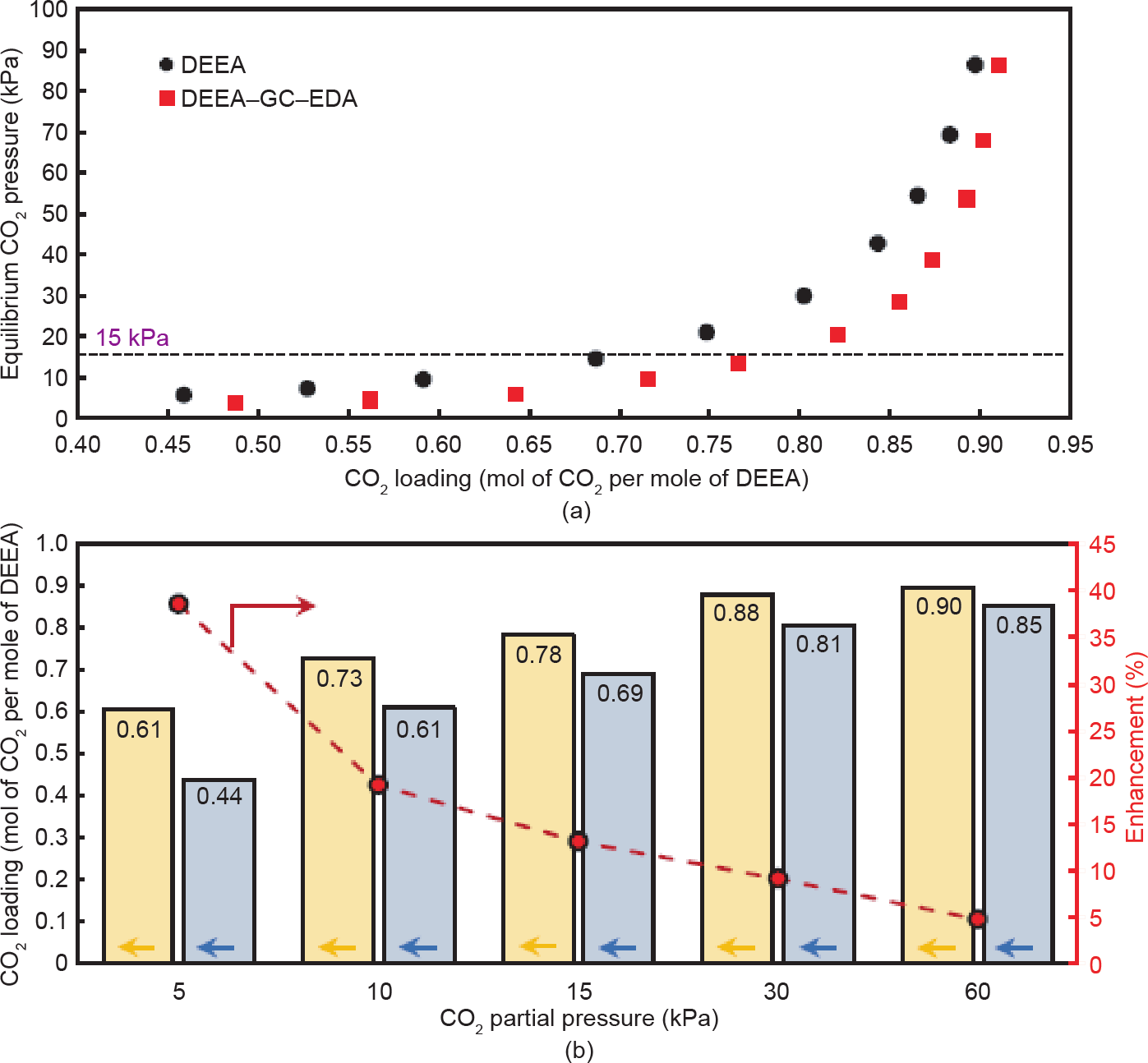
图8. (a)CO2吸收等温线;(b)传统的DEEA水溶液(灰色)和DEEA-GC-EDA纳米流体(黄色)的平衡CO2负载量和增强比。
为了测试纳米多孔NCP-GC-EDA作为新引入的CO2 促进剂的稳定性,我们在三个连续的吸收-解吸循环中,实验测量了CO2吸收速率、与DEEA水溶液相比的CO2 速率增强情况以及DEEA-GC-EDA纳米流体的再生效率。如图9(a)所示,在第一个循环期间,再生效率降至96.19%,导致CO2吸收速率从50.70 kPa·min–1(相对原始DEEK水溶液提高38.6%)降低到48.77 kPa·min–1 (提高了32.88%)。这种减少是因为胺位点的部分变形(或饱和)及表面上CO2和伯胺基团之间不可逆的物理吸收和化学反应而导致的GC结构阻塞。在类似的趋势中,第二周期的CO2吸收速率和再生效率逐渐下降至47.89 kPa·min–1和94.46%,第三周期达到 48.06 kPa·min–1和94.79%。观察到第二个和第三个循环具有相似的再生效率值,所以假设DEEA-GC-EDA纳米流体的CO2吸收速率在连续三个循环后趋于稳定。分析数据表明,DEEA-GC-EDA纳米流体的CO2循环吸收速率仅比其初始CO2吸收速率低5.2%(50.7 kPa·min–1至 48.06 kPa·min–1),这表明纳米多孔NCP-GC-EDA在循环 CO2中具有高性能的吸收-解吸运作过程。因此,使用叔胺水溶液和NCP-GC-EDA促进剂可从烟气中大规模吸收CO2。
《图9》
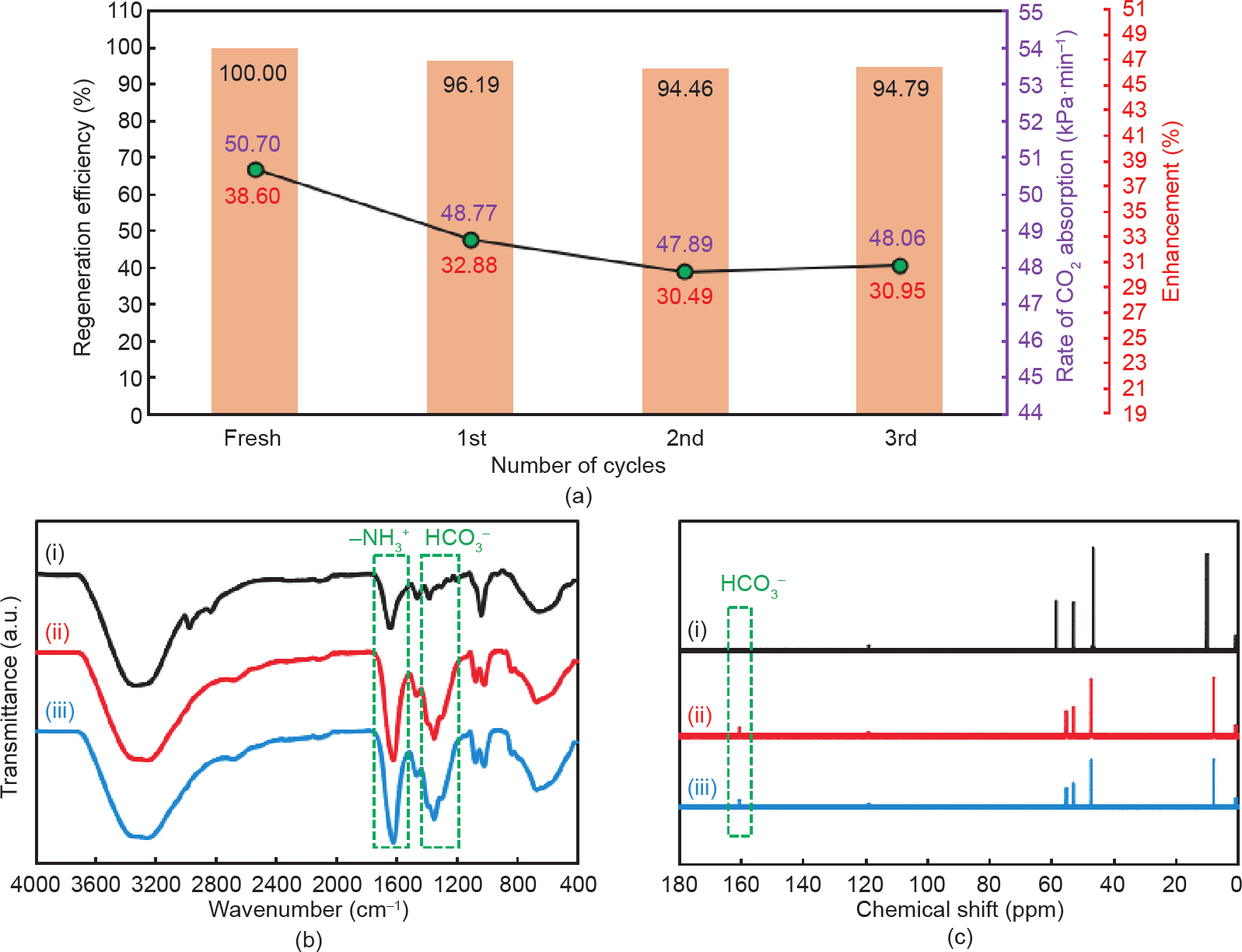
图9. (a)连续三个循环的DEEA-GC-EDA纳米流体的CO2吸收率、再生效率和增强比;新鲜DEEA水溶液在吸收CO2之前(i)和吸收CO2之后(ii)的FT-IR(b)和13C NMR(c),以及DEEA-GC-EDA纳米流体(iii)在吸收CO2之后的FT-IR(b)和13C NMR(c)。
为了研究微孔NCP-GC-EDA在DEEA水溶液中促进吸收CO2的机理,将CO2饱和的DEEA-GC-EDA纳米流体在10 000 r·min–1下离心15 min,剩余的褐色上清液通过FT-IR和NMR图谱表示。如图9(b)和(c)以及附录A中的图S7(b)所示为饱和CO2的DEEA-GC-EDA 纳米流体、不使用任何促进剂的CO2饱和的DEEA溶液和淡水DEEA溶液(在吸收CO2之前)的FT-IR、13C NMR和1H NMR谱图。对不同样品进行FT-IR光谱分析后进行比较,可以发现,DEEA水溶液和DEEA-GCEDA纳米流体都包含相似的物质。在1350~1360 cm–1和 1560~1625 cm–1区域吸收带强度的增加与CO2吸收过程中碳酸氢盐和质子化胺的形成有关[41]。不同样品的1H NMR和13C NMR光谱显示的结果与FT-IR相似,确认了碳酸氢盐是溶液中存在的唯一含CO2的物质。值得注意的是,两种表征方法都强调在吸收CO2期间溶液中未形成氨基甲酸酯。这一发现表明,使用碳质NCP-GC-EDA 促进剂不会影响叔胺溶液中的CO2吸收机理,并且该反应受碳酸氢盐的形成反应所控制。因此,可以推断出,纳米多孔NCP-GC-EDA可以通过物理吸收的CO2分子与接枝多胺的伯/仲胺基团之间的氨基甲酸酯形成反应,从而提高叔胺的CO2吸收速率和平衡CO2吸收能力。
《4. 结论》
4. 结论
总而言之,本文利用具有多胺官能团的新型纳米多孔速率促进剂,以提高水性叔胺中CO2的吸收速率。为了评估孔隙结构和胺官能团的作用,我们采用了GC 和GS碳结构,并分别通过低相对分子质量的EDA和高相对分子质量的PEI分子后功能化。通过FT-IR、XPS、 TGA、Raman、SEM和元素分析对所有合成的NCP进行了全面表征,以评估促进剂的结构特性。在DEEANCP纳米流体水溶液中CO2吸收的性能研究证实,包含EDA接枝GC结构的DEEA-GC-EDA纳米流体在所有制备的纳米流体中具有最强性能。具体如下,在典型的DEEA水溶液中仅使用0.1 wt%的NCP-GC-EDA促进剂就可以成功地将CO2吸收率从36.8 kPa∙min–1提高到 50.7 kPa∙min–1(增强率为38.6%),整个反应的CO2吸收率从每摩尔DEEA吸收0.69 mol提高至0.78 mol(提高了13.2%)。此外,本文研究了NCP-GC-EDA浓度对 CO2吸收速率的影响,并证明0.1 wt%是DEEA水溶液中NCP的最佳浓度。为了评估NCP-GC-EDA的重复使用性,本文对DEEA-GC-EDA纳米流体的循环CO2吸收率进行了测量,在三个连续的CO2吸收-解吸循环中仅显示出5.2%的减少,吸收速率从50.7 kPa·min–1降低至 48.06 kPa·min–1。此外,在DEEA-GC-EDA纳米流体中吸收CO2前后,基于FT-IR和NMR光谱分析,本文提出了NCP-GC-EDA促进剂的增强机制。总的来说,微孔 NCP-GC-EDA可以作为一种可行的促进剂,能够有效克服叔胺溶液的缺点,从而提高CO2的吸收速率并促进其大规模利用。
《Acknowledgements》
Acknowledgements
The authors would like to acknowledge the University of Melbourne for the Melbourne Research Scholarship, infrastructural support, and financial resources provided for this project. The authors also acknowledge the support of the Bio21 Advanced Microscopy Facility (the University of Melbourne) and Melbourne TrACEES Platform (Trace Analysis for Chemical, Earth and Environmental Sciences) for the technical support, data analysis, and expert advice.
《Compliance with ethics guidelines》
Compliance with ethics guidelines
Masood S. Alivand, Omid Mazaheri, Yue Wu, Geoffrey W. Stevens, Colin A. Scholes, and Kathryn A. Mumford declare that they have no conflict of interest or financial conflicts to disclose.
《Appendix A. Supplementary data》
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.eng.2020.05.004.











 京公网安备 11010502051620号
京公网安备 11010502051620号




