
《1、 引言》
1、 引言
结直肠癌是最常见的癌症之一,也是全球第三大癌症死亡原因。许多研究已经证实了肠道菌群和结直肠癌发展之间的联系,其中大多数研究是基于生物信息学分析[1‒3]。16S rRNA基因的扩增子测序和宏基因组测序是研究肠道微生物组的主要方法,已被广泛用于鉴定可能的结直肠癌致病菌,以及寻找结直肠癌潜在的诊断标志物[4]。当细菌被证明与结直肠癌相关后,就会用细胞或者动物实验来验证这种相关性。通过测序技术来确定微生物的生理状态和功能是比较困难的;此外大多数研究中用到的菌株通常是从保藏中心购买的,而不是分离自临床患者[5‒6]。
同种细菌的菌株之间也存在很大的表型差异。比如,菌株水平的差异会影响膳食化合物的代谢,如半乳糖和不易消化的纤维[7‒8]。细菌介导的药物代谢也可能因菌株而异,从而影响药物的疗效和毒性[9]。尽管许多技术已经被用于从数千个与人类相关的细菌宏基因组中提取物种和亚种水平的信息,但使用不依赖于培养的方法在菌株水平上准确区分这些特征是极其困难的[10‒11]。培养组学是一种使用多种培养条件来获得细菌,并采用MALDI-TOF质谱和16S rRNA测序来鉴定细菌的方法[12]。该技术已成为扩展我们对微生物组理解的关键方法之一,同时也是获得目标菌株,并在菌株水平上研究微生物群与宿主之间相互作用机制的重要方法。如最近的研究显示,Sorbara等[13]从人类样本中培养了273个毛螺科菌株,包括11个属和27个种,并证明了它们在种间和种内水平上的多样性,证实了微生物群在菌株水平上研究的意义。
在肠道菌群和结直肠癌相关性研究中,样本大多数来自于较容易获得的粪便样本,但是肠黏膜相关细菌可能在微生物群与宿主的相互作用中发挥更重要的作用。研究显示,通过16S rRNA扩增子测序证实了黏膜微生物群与结直肠癌更为相关,并发现黏膜中肠道微生物群的多样性和组成与粪便中微生物群的多样性和组成不同[14‒15]。最近的一项研究指出,肠黏膜样本可能更适合研究微生物群与宿主之间的关系[16]。为了进一步证明黏膜相关细菌在菌株水平上的差异,我们通过培养组学,从22例结直肠癌患者的44份肠黏膜标本(包括癌症黏膜和癌旁黏膜)中分离出158株大肠杆菌,利用二代测序技术对其进行全基因组测序,并在细胞和动物水平上进行细胞因子检测。然后,进一步分析与毒力相关的基因分布和代谢途径,以寻找功能差异的可能机制,证实了不同菌株的大肠杆菌可表现出较大的基因组变异和功能差异。
《2、 材料和方法》
2、 材料和方法
《2.1 样本》
2.1 样本
纳入研究的22例结直肠癌患者来自北京世纪坛医院。样本入选标准:①50~80岁的结直肠癌患者;②既往未接受过放化疗;③符合手术治疗标准。取2 cm2的结直肠癌组织或癌旁组织,在2 mL磷酸盐缓冲液(PBS)中完全研磨。在预定的培养条件下分离培养组织中的菌株。
《2.2 培养组学》
2.2 培养组学
采用培养组方法对细菌进行分离和鉴定。将黏膜组织置于无菌PBS中进行研磨;所有样品均在基础培养条件下培养(有氧或厌氧,37 ℃,YCFA固体培养基);当平板上出现单克隆时,挑取单克隆并转移至24孔板中的液体培养基中进行液体扩增。扩增产物加入甘油进行冻存,同时对扩增产物进行固体培养基三区划线。待三区划线长出单克隆后,用AutOF MS1000光谱仪(Autobio,郑州)鉴定细菌种类。如果MALDI-TOF MS不能准确鉴定菌落,则通过16S rRNA基因测序鉴定分离株。16S rRNA基因测序由擎科生物科技公司(北京)完成。
《2.3 生物信息学分析》
2.3 生物信息学分析
《2.3.1. 大肠杆菌的分类》
2.3.1. 大肠杆菌的分类
大肠杆菌基因组测序由诺禾生物技术公司(北京)完成。序列数据已上传至GenBank (Bioproject pRJNA608078)。我们使用了克隆框架软件[17]。基于8个管家基因(总共4095个核苷酸)的序列对大肠杆菌进行分析和分类[18]。我们使用Snippy Pipeline v4.3.8 (https://github.com/tseemann/snippy)进行SNP调用,使用E. coli STR K-12子代MG1655(登录号:U00096)作为参考基因组。位于重复区域的SNPs被排除在系统发育分析之外。用TRF V4鉴定重复区域,并通过BLASTN进行自排列。使用TreeBest软件(http://treesoft.sourceforge.net/treebest.shtml)将全基因组SNPs用于构建邻接树,并使用GGTree对所有系统发育树进行可视化。
《2.3.2. 大肠杆菌基因分析》
2.3.2. 大肠杆菌基因分析
我们用SPAdes进行了重新组装。用Prokka对组件进行基因注释,并在Roary中使用注释结果(GFF3文件)来识别泛基因组并生成基因存在/不存在矩阵。我们使用eggNOG-Mapper来注释泛基因的直源同源群集(COG)分类。
《2.4 Transwell实验》
2.4 Transwell实验
Transwell平板是一种嵌套结构,它将孔分为上室和下室。嵌套中间的膜具有不同孔径规格。我们选择0.4 μm的孔径以确保细菌产物可以通过膜而细菌不能通过。将THP-1细胞以每孔106个细胞(106个细胞∙mL-1)的接种密度在12孔Transwell平板(Thermo Fisher Scientific)的下室中接种。然后细胞加入添加终浓度为100 μg∙mL-1的佛波醇12-肉豆蔻酸酯13-乙酸酯(PMA)(Thermo Fisher Scientific)的培养基中。在37 ℃、5% CO2的湿润空气中孵育48 h,以刺激细胞分化为巨噬细胞。随后,将大肠杆菌菌株离心后除去细菌培养基,重悬于细胞培养基中。最后,将不同的大肠杆菌菌株以MOI=10加入Transwell平板的上室(每孔107个)。使用RPMI 1640培养基作为阴性对照。在37 ℃、含5% CO2的湿润空气中孵育3 h后,收集细胞培养基。THP-1细胞(人白血病单核细胞)购自北纳生物技术公司(美国),细胞培养基为添加10%牛血清(Cleson Scientific,中国)的RPMI 1640(Thermo Fisher Scientific)。
《2.5 动物实验》
2.5 动物实验
雌性BALB/c小鼠(8周龄)购自维通利华生物公司(北京)。将小鼠随机分为5组,每组5只小鼠:A1、A2、D1、D2和B1。小鼠适应环境一周后,在其饮水中加入广谱抗生素:1 g∙L-1氨苄青霉素、1 g∙L-1硫酸新霉素、1 g∙L-1甲硝唑和0.5 g∙L-1万古霉素[19]。喂食后,每只小鼠立即用109 cfu∙mL-1大肠杆菌灌胃,每周两次,共4周,每周尾部采血一次。空白对照组给予等体积无菌PBS。
《2.6 细胞因子检测》
2.6 细胞因子检测
收集Transwell装置中THP-1细胞的培养上清液,并用Bio-Plex 200®系统(Bio-Rad, USA)进行测定。
《2.7 分析和统计》
2.7 分析和统计
对多个组的细胞因子含量进行比较。首先确认每组数据的正态性和方差齐性。如果数据呈正态分布且显示方差齐性,则使用方差分析(ANOVA),而如果数据不呈正态分布或显示方差异质性,则使用秩和检验(P < 0.05)。采用Pearson´s χ2检验比较两组间致病基因的分布情况。两组总样本数定义为n,理论频数定义为t,当n ≥ 40且所有t ≥ 5时,采用Pearson´s χ2 检验,当n < 40或t < 1时,采用Fisher确切概率检验(P < 0.05)。
《3、 结果》
3、 结果
《3.1 结直肠癌黏膜来源的大肠杆菌菌株》
3.1 结直肠癌黏膜来源的大肠杆菌菌株
黏膜是与肠道疾病最相关的组织,因此我们使用培养组学方法分离和培养了44个黏膜样本。从结果中可以看出,大肠杆菌是从结直肠癌和邻近黏膜中分离出的最常见的菌种,它可以从所有44个样品中分离出来(附录A中的图S1)。所有大肠杆菌菌株和患者信息如表S1所示。
《3.2 大肠杆菌在结直肠癌患者肠道微生物群中的遗传多样性》
3.2 大肠杆菌在结直肠癌患者肠道微生物群中的遗传多样性
共生大肠杆菌的基因结构是由多种宿主和环境因素决定的。决定其毒力的因素可能反映了其对共生环境的适应。我们选择了158株大肠杆菌进行全基因组测序。在这些菌株中,来自同一患者的菌株用于评估宿主内的多样性,来自不同患者的菌株用于宿主间的比较。利用Clonal Frame软件对大肠杆菌进行分类分析[17,20],菌株被分为5个类群,即A、D、B1、B2和F(图1)。但是来自癌组织和癌旁组织的大肠杆菌菌株在系统发育树上没有明显区分,表明来自两种组织的菌群之间不存在基因型差异。
《图1》
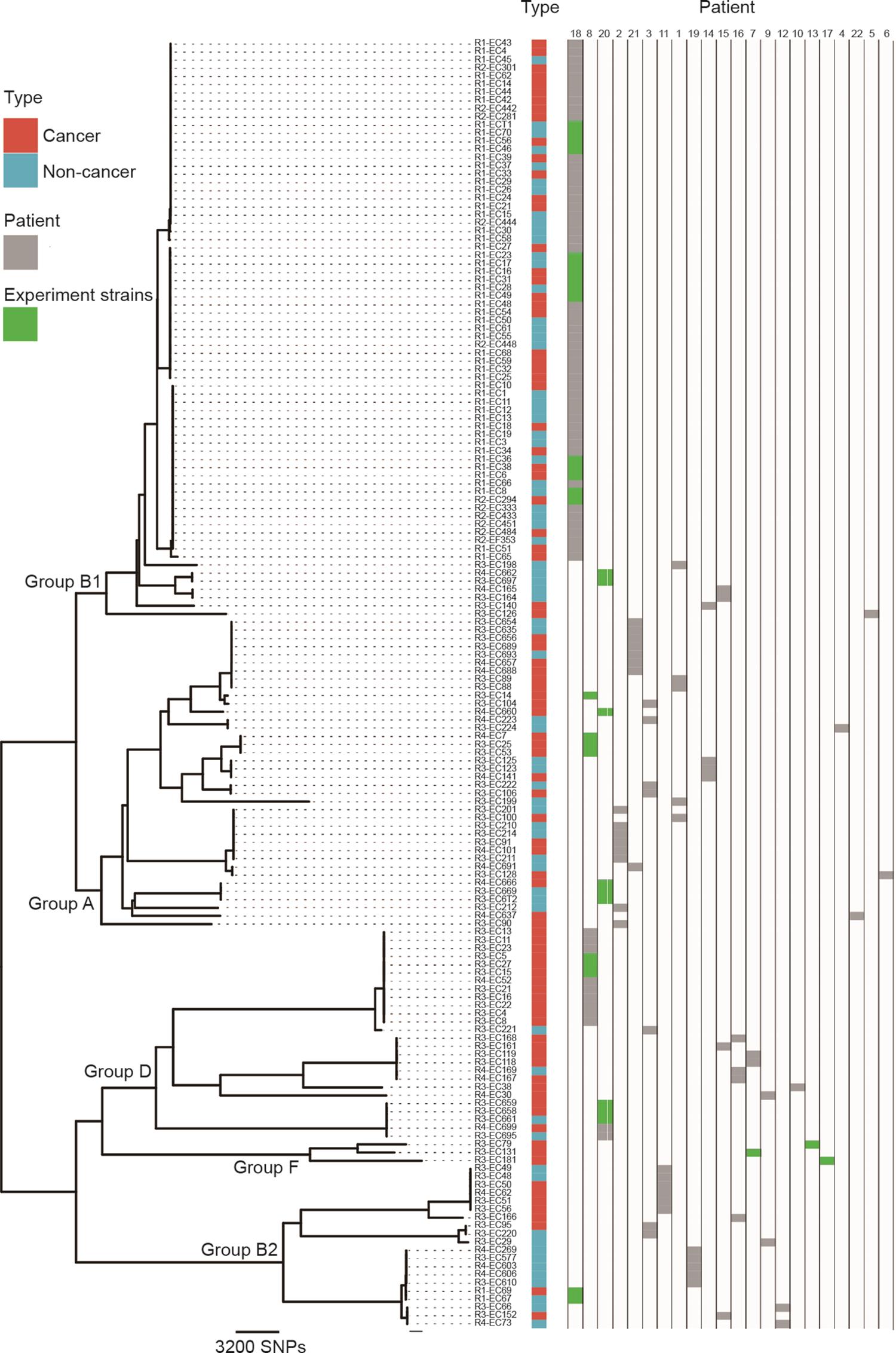
图1 结直肠癌相关大肠杆菌的系统发育树。对来自22名患者的158株大肠杆菌进行了分析。这些菌株可分为5个主要的系统发育群:A、B1、B2、D和F。红色表示来自癌组织的菌株,蓝色表示来自非癌组织的菌株。对于进一步的Transwell实验菌株用绿色标注。
《3.2.1. 宿主内多样性》
3.2.1. 宿主内多样性
早期一项研究表明,在任何时候,一个人通常携带一种占优势的微生物群菌株,该菌株占分离出的菌落的一半以上,其他菌株也以不同的水平存在[21]。在本研究中,我们注意到一些患者携带一个优势系统类群。例如,在患者18中,大多数菌株属于种系群B1,然而,基于单核苷酸多态性(SNP)分析,它们可以进一步区分为不同的分支,表明这些菌株在基因组水平上的差异。在患者2中,仅分离出来自系统类群A的菌株。相反,在患者3中,菌株被分配到三个系统类群,没有优势系统类群。
《3.2.2. 宿主间多样性》
3.2.2. 宿主间多样性
从结果中可以看到,宿主之间菌株水平的细菌多样性很高,我们没有检测到任何两个患者有完全相同的菌株。这些结果进一步加深了对细菌菌株功能的理解,这对于细菌在疾病发展和微生态研究中的精确机制至关重要。随后,我们采用免疫细胞实验和动物实验来进一步评估不同种系代表株的细胞因子诱导效果。
《3.3 大肠杆菌不同菌株对THP-1细胞的影响》
3.3 大肠杆菌不同菌株对THP-1细胞的影响
由于肠道微生物群在正常状态下不与免疫细胞直接接触,因此肠道细菌对免疫细胞的作用主要是通过其代谢产物介导的。因此,使用Transwell方法来研究不同大肠杆菌菌株对免疫细胞的影响[22]。根据大肠杆菌类群分类,共选择36个代表性菌株(来自6名患者)用于实验。Transwell平板分成上室和下室,首先将THP-1细胞(人白血病单核细胞)加入下室,然后用PMA诱导细胞48 h以诱导其分化为巨噬细胞,最后将相应的大肠杆菌菌株加入上室(感染复数[MOI] = 10)。孵育3 h后,收集细胞培养基以测定白细胞介素1β(IL-1β)、肿瘤坏死因子α(TNF-α)和白细胞介素6(IL-6)的水平[图2(a)]。
《图2》
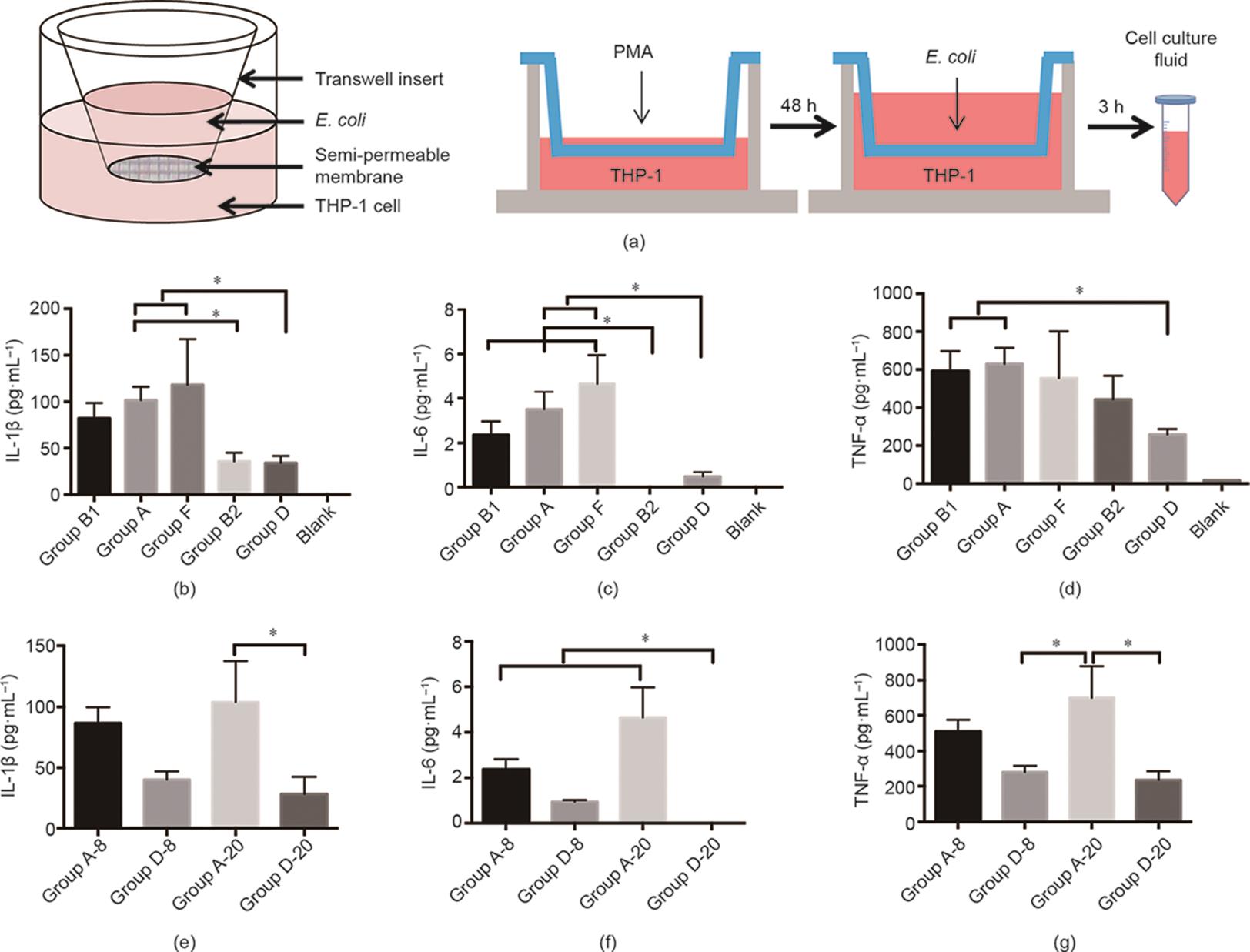
图2 (a)Transwell实验设计;(b)各组细胞培养基中IL-1β含量的比较;(c)各组细胞培养基中IL-6含量的比较;(d)各组细胞培养基中TNF-α含量的比较;(e)细胞培养基中IL-1β含量的组内比较;(f)细胞培养基中IL-6含量的组内比较;(g)细胞培养基中TNF-α含量的组内比较。*P ≤ 0.05。
首先,我们比较了来自不同种类群的大肠杆菌菌株对巨噬细胞分泌这三种细胞因子的影响[图2(b)~(d)]。结果表明,5个类群之间存在显著差异。D群菌株刺激后细胞因子的分泌量低于其他群菌株,而A群菌株刺激后细胞因子的分泌量始终高于D群菌株。我们还研究了样品来源和样品分类是否影响巨噬细胞分泌细胞因子。结果表明,即使菌株来自同一患者,不同种系的菌株之间也存在显著差异;但即使样本来自不同的患者,细胞因子诱导效应在同一系统类群中也是相似的[图2(e)~(g)]。总之,菌株的种系对巨噬细胞分泌细胞因子的影响比菌株的来源更明显。
《3.4 不同大肠杆菌菌株对小鼠的影响》
3.4 不同大肠杆菌菌株对小鼠的影响
由于在细胞实验中A群和D群菌株之间存在显著差异,我们从系统发育树分支中选择了两株大肠杆菌进行动物实验分析。我们将四个菌株分别命名为A1、A2、D1和D2,以及空白对照组BL。我们首先用联合抗生素喂养小鼠1周以清理其肠道微生物。在抗生素处理后,用相应的大肠杆菌菌株每周灌胃小鼠两次,持续4周,并且在每周第二次灌胃两天后收集血样[图3(a)]。在实验过程中,各组之间未观察到体重差异(图S2)。实验结束时,与对照组相比,四个实验组的结直肠病理无明显变化(图S3)。每周检测一次血清中IL-1β、TNF-α和IL-6的含量[图3(b)~(d)]。在第1周,细胞因子的变化与细胞实验的结果一致,而在第2周和第3周,4个实验组和对照组之间没有观察到差异。但在第4周,血清中细胞因子的浓度与第1周相反,用D组菌株刺激后的细胞因子浓度显著高于用A组菌株刺激后的细胞因子浓度。
《图3》
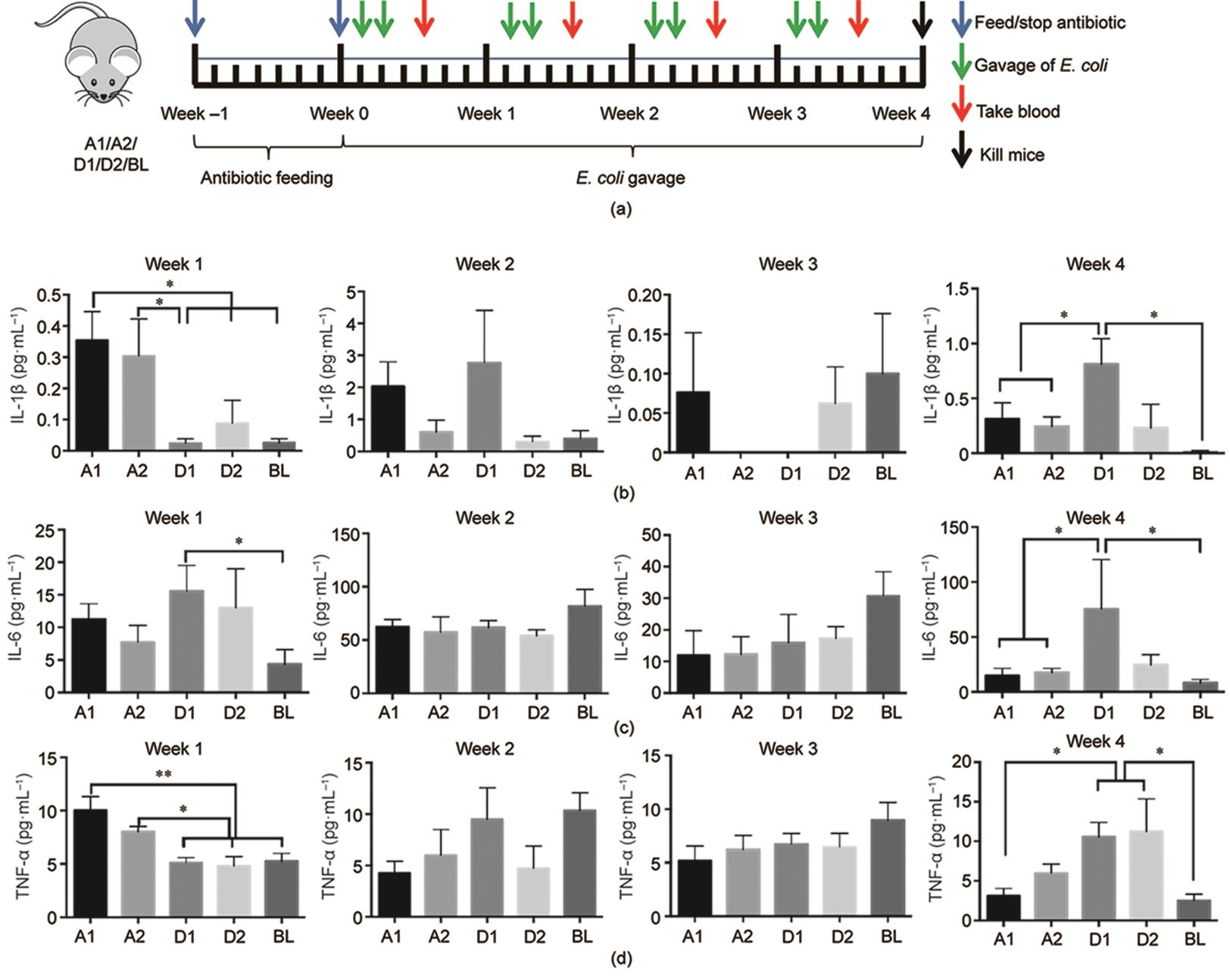
图3 (a)小鼠实验设计;(b)灌胃后1~4周小鼠血清中IL-1β含量的比较;(c)灌胃后1~4周小鼠血清中IL-6含量的比较;(d)灌胃后1~4周小鼠血清中TNF-α含量的比较。*P ≤ 0.05;**P ≤ 0.01。
《3.5 大肠杆菌A群和D群菌株的遗传分析》
3.5 大肠杆菌A群和D群菌株的遗传分析
为了探索两个大肠杆菌系统群之间对于细胞因子诱导差异的可能机制,我们进一步分析了细胞实验中使用的14个菌株(6个来自系统群D,8个来自系统群A)的基因组差异,以了解毒性相关的基因分布和代谢途径。我们在谱系特异性核心基因中发现了33 248个SNPs,在谱系特异性辅助基因中发现了348个SNPs [图3(a)和表S2]。当我们将SNP相关基因定位到京都基因和基因组百科全书(KEGG)通路时,两个系统类群之间有37条显著不同通路(表S3)。在348个基因中,278个基因特异性地存在于系统类群D中,70个基因特异性地存在于系统类群A中。这些基因大部分与细菌代谢有关,少数是与细胞侵袭和黏附有关的毒力相关基因。与核心基因相关的途径包括氨基酸、核苷酸、碳水化合物和无机离子的转运和代谢。与辅助基因相关的途径仅涉及碳水化合物转运和代谢(图S4)。
前期研究显示,在大肠杆菌中,有100多个基因与毒力有关[23‒31]。我们筛选了103个毒力相关基因或基因组岛,并比较了A和D类群菌株中的这些基因。然后,我们计算了这些基因在每个系统类群中的比例(表S4)。在图4(b)~(d)中展示了显著不同的基因,包括测序菌株[图4(b)]、细胞实验中使用的菌株[图4(c)]以及在动物实验中使用的菌株[图4(d)]。14个基因(chuA、ipaH、 fmlA、fimA、fimC、ecpA、ecpD、kpsS、ydeR、FimH、iss、aufC、lpfA和stfD)在D群中的频率高于A群(P ≤ 0.025),12个基因(yfcV、ChiA、elfC、stcD、papC、iucC、iucD、iutA、iha、sat、papF和papGII)在A群菌株中比在D群菌株中更普遍(P ≤ 0.050)。据报道,系统类群D中的几个基因(如chuA、ipaH、fmlA)与黏附和侵袭有关,而系统类群A中的yfcV与黏附有关。
《图4》
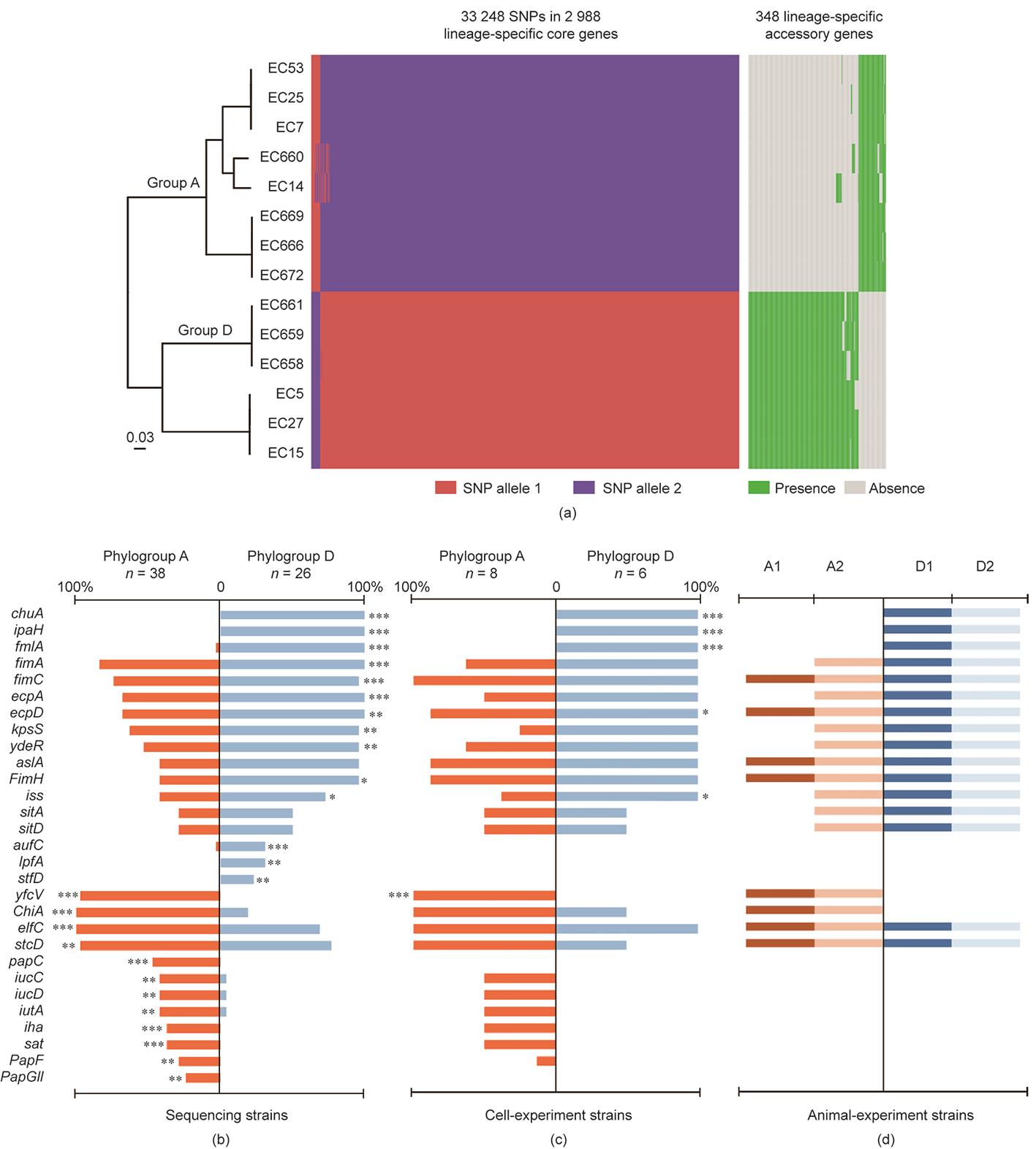
图4 (a)谱系特异性核心基因和谱系特异性辅助基因的单核苷酸多态性(SNPs)在A群和D群之间的比较。(b)~(d)致病基因在A群和D群中的分布:(b)所有测序菌株;(c)仅用于细胞实验的菌株;(d)仅用于小鼠实验的菌株。数字表示含有基因菌株数量相对于该类群的菌株总数的百分比。图中展示的是有统计学显著差异的基因。*P ≤ 0.05;**P ≤ 0.01;***P ≤ 0.001。
虽然我们没有发现来自不同患者的任何相同菌株,但我们在来自不同患者的菌株中观察到相同的毒力基因谱(图S5)。例如,分别来自患者21和患者4的菌株R4-EC223和R3-EC224具有相同的已知毒力基因谱。又比如,菌株R3-EC201和R3-EC100不仅来自不同的患者,而且来自不同的黏膜部位(癌症和邻近组织),也具有相同的毒力基因谱。
《4、 讨论》
4、 讨论
目前,大多数关于肠道细菌和结直肠癌的报道都采用宏基因组学方法鉴定相关细菌,然后使用美国典型培养物保藏中心(ATCC)的菌株进行进一步验证。例如,具核梭杆菌(Fusobacterium nucleatum)被发现与结直肠癌相关,菌株ATCC 23726和25586被用于在无菌小鼠中诱导肿瘤[5,32]。虽然AOM在肠黏膜损伤后成功诱导了肿瘤,但使用结直肠癌患者的原始菌株进行验证实验,这可能更有助于理解靶细菌在癌症发展中的致病作用。
在本研究中,分离自结直肠癌患者黏膜组织的大肠杆菌菌株,从基因组和功能上均体现了菌株差异。大肠杆菌是人体肠道中常见的一种细菌,有些会引起胃肠道疾病[33],也会引起泌尿系统[34]、神经系统感染[35]。同时有宏基因组数据也表明克罗恩病患者个体中存在多种大肠杆菌菌株[36]。根据基因组分类结果,158株大肠杆菌被分为A、D、B1、B2和F共5个类群。我们发现,来自不同患者的大肠杆菌菌株属于不同的类群,表明不同宿主间,肠道微生物群中的细菌菌株具有高度多样性。随后,我们进一步找出了这些类群的功能差异。
细胞因子是反映肿瘤患者免疫反应的重要指标。IL-1β、TNF-α和IL-6是在癌症中表达变化显著的三种细胞因子,它们的水平甚至可能与肿瘤发展的阶段相关[37‒38]。因此,使用Transwell实验,我们首先检测了来自不同类群的不同大肠杆菌菌株对巨噬细胞的刺激情况。结果显示,不同类群间差异明显,A群和D群诱导的细胞因子水平差异最为显著。如果来自同一患者的菌株属于不同的系统群,这些菌株在诱导免疫细胞分泌细胞因子方面是否存在差异?我们比较来自患者8和患者20的A群和D群中的菌株。结果显示来自同一种类群的大肠杆菌菌株对细胞因子的诱导没有差异。但是,大肠杆菌不同类群的菌株刺激免疫细胞后,其分泌的细胞因子水平明显不同(例如,来自同一患者的A-20和D-20诱导的IL-1β和TNF-α分泌有显著性差异)。这些结果提示,对于肠道微生物群的研究要在菌株水平展开,以准确了解细菌菌株在肠道中的作用。
接下来,我们用来自A群和D群的菌株刺激小鼠,来观察是否可以在动物模型中检测到类似的结果。在细菌刺激之前,所有的小鼠都接受了抗生素治疗,以清除它们的肠道微生物群[19]。菌株灌胃一周后,A、D两组小鼠血清IL-1β和TNF-α水平有显著差异,与细胞实验结果一致。在接下来的两周内,用不同菌株刺激的小鼠,其血清细胞因子分泌没有差异。然而,在灌胃后的第4周,与A组相比,D组的菌株再次在小鼠中引起显著的细胞因子分泌。这是一个有趣的表型结果,我们试图在基因水平上对其进行解释。我们首先分析了A群和D群中存在和缺失的基因。总共鉴定了352个类群特异性辅助基因,这些基因主要存在于D群中。在A群中,大多数大肠杆菌菌株是产肠毒素性大肠杆菌(ETEC),其通过表面蛋白和可能的其他表面结构附着于宿主上皮而引起疾病[23]。然而,黏附侵袭性大肠杆菌(AIEC)与炎症性肠病有关,可产生引起肠道炎症和病变的毒素[27],主要分布在类群D和B2中。研究表明,表达yfcV基因的大肠杆菌比不表达yfcV基因的大肠杆菌更容易黏附到黏膜上,从而引起炎症。yfcV是一种黏附基因,编码假定的伴侣蛋白的主要亚基[39]。yfcV基因在A组中的表达显著高于D组,这可能与A组在细胞实验中和刺激小鼠后第一周血清中的细胞因子表达显著升高有关。然而,随着感染时间的延长,具有比A群更多毒力基因的D群可能诱导宿主产生更多的细胞因子。早期研究也提到,AIECs会有多种毒力因子来促进其对细胞的侵袭[40]。
除了基因缺失,SNPs的差异也会在菌株水平上影响细菌的功能。例如,与黏附相关的S层糖蛋白的gmhB基因在系统发育D群中表达被中断[41]。又比如,A群中的yghJ基因(也称为SSIE)框内缺失破坏,可能导致其失活。研究表明,yghJ基因与细菌分泌系统和生物膜形成有关,触发NF-κB和MAP激酶信号通路,从而诱导炎症因子的表达[42‒43]。因此,A群和D群中的SNPs也可能导致表型差异。
有研究表明,不同的细菌菌株对人类健康的影响不同。目前人类肠道中大多数细菌物种的活体分离菌株及基因组信息仍然有限,特别是在菌株水平的研究更为有限。此外,大多数用于菌株水平研究的菌株来自不同的个体,而我们对来自同一人体的菌株多样性的了解非常有限。一些研究指出,同一物种的菌株之间存在功能差异,这可能会影响人类健康[44‒45]。因此,使用培养法从结直肠癌患者的肿瘤黏膜及其邻近组织中分离出黏膜相关细菌,这可以为未来的功能研究提供了结直肠癌相关的肠道微生物菌库。本研究中对大肠杆菌菌株进行分离培养、测序,及分析它们对细胞和宿主的功能,进而发现了可能的作用机制(图5),为在菌株水平上进行深入的筛选、分析和验证提供了研究基础。
《图5》

图5 菌株水平大肠杆菌对宿主的影响及机制。











 京公网安备 11010502051620号
京公网安备 11010502051620号




