

《1、 概述》
1、 概述
水泥是建筑行业最常用的黏合剂,似乎没有替代材料可以很快地取代它。在水泥水化产物中,水合硅酸钙(C-S-H)是主要成分,它主要决定硬化水泥和混凝土的力学性能和耐久性能。水泥基性能的改进需要在基本尺度上对凝胶体系有深刻的理解。因此,可以预期水泥基研究从宏观性质到“原子级行为”的转变可能会解开隐藏已久的有用信息[1]。在原子水平上对水泥基材料进行分子模拟,最终将使工程师确保以最佳的力学性能和耐久性的方式设计水泥混合物,从而实现环境可持续发展。
混凝土是一种纳米结构、多相和多尺度的材料(即其尺寸从纳米到微米到宏观不等),如图1所示。它具有晶相和非晶相以及相界面。一般来说,纳米尺度的特性决定了宏观尺度的特性,从而决定了混凝土在工作载荷下的宏观性能[2]。控制混凝土大部分性能的最关键问题在于从水化开始了解水泥浆的化学性质及其相关的物理变化,如凝固和硬化。水泥的水化是一个复杂的化学过程,涉及溶解、扩散、成核和生长,它与系统的化学和物理力学变化有关[3‒7]。这一过程远未被人们完全理解,因为它们可能以非单调的方式经历复杂的相互作用[8]。因此,大多数用于研究水泥水化动力学的实验方法都是基于监测微观结构形成的净速率[9]。然而,这些实验都没有详尽地理解水化机制,而是有效地比较和表征了影响参数,如水/水泥(w/c)比、水泥成分、温度、湿度等。因此,需要了解纳米尺度的水化机制,以从这种廉价且常用的材料中获得最佳性能。
《图1》

图1 水泥体系多维度描述示意图。DEM:离散元法;FEM:有限元法;XFEM:扩展有限元法;XRD:X射线衍射;AFM:原子力显微镜;SEM:扫描电子显微镜;TEM:透射电子显微镜;ITZ:界面过渡区。
自从20世纪50年代初期以来,分子动力学(MD)模拟和计算能力不断发展,人们在建筑材料的参数性能和结构模拟方面取得了一些突破。除了水泥,MD模型还被用于研究其他土木工程材料,包括黏土[10‒12]、沥青[13‒14]、木材[15‒17]等。
随着计算能力的无限提升,如超级计算机的使用,现在可以通过分子模拟来评估在建筑行业中使用纳米工程的可能性。然而,迄今为止,对使用分子模拟研究水泥基材料相互作用的详细综述依然不足。因此,本文旨在通过全面调查目前该领域进行研究的范围和水平,科学地讨论关键结果和发现,以期为未来的研究提供建议,从而填补这一知识空白。
本文首先简要概述了原子建模和模拟,其次讨论了水泥体系模拟的新方向(图2)和水泥的性能,重点关注其结构-性能的关系,还从基本的自下而上角度讨论了水泥与水和离子的相互作用。希望本文进行的讨论能够启发研究人员使用分子水平模拟来预测水泥基材料的宏观行为。
《图2》

图2 水泥基材料的分子模拟。术语C、S、A和F在水泥化学中分别代表CaO、SiO2、Al2O3和Fe2O3。
《2、 分子模拟》
2、 分子模拟
理论方法与计算技术的结合促进了材料的分子模拟。分子建模成为不可或缺的工具,并已被广泛应用于化学、材料科学、药理学、生物学、能源等多个领域。近年来,由于纳米工程和现代建筑材料的研究取得了进展,在水泥基材料中利用分子建模得到了更多的关注。为了与这一趋势保持一致,分子模拟提供了独特而有用的方面,例如,可以更好地理解材料及其在基本尺度上的相互作用以及测试独立因素对材料行为的影响[18]。
分子建模主要基于两种方法:量子力学(QM)和经典力学(CM)。在QM方法中,薛定谔方程通过使用从头计算或半经验方法求解。QM考虑的是系统的电子状态,因此计算成本高且要求严格。另一方面,使用CM方法的数值建模依赖于经典物理学(即非量子并基于经典牛顿力学)。它将原子视为质量粒子,其键表示为弹簧。因此,这些原子间的键可以模拟为拉伸、弯曲和扭曲(附录A中的图S1和图S2)。
经典分子建模主要涉及三种技术:分子力学(MM)、分子动力学(MD)和蒙特卡罗(MC)模拟(附录A中的表S1)。MM技术依赖于传统的CM来模拟分子系统。在这种方法中,原子间的相互作用是基于势函数(或通常称为力场)获得的。MC模拟基于吸附过程的随机采样[19]。
《3、 硅酸盐水泥相的分子模拟》
3、 硅酸盐水泥相的分子模拟
《3.1 无水相》
3.1 无水相
普通硅酸盐水泥(OPC)是一种石灰基材料。它基本上由60%~67% CaO、17%~25% SiO2、3%~8% A12O3、0.5%~6% Fe2O3、0.5%~4% MgO、0.3%~1.2%碱和2%~3.5% SO3 [20]构成。然而,水泥成分通常用矿物成分来表示,即硅酸三钙(C3S)、硅酸二钙(C2S)、铝酸三钙(C3A)和铁铝酸四钙(C4AF);由于它们与相应的矿物相似,它们有时分别被称为阿利特(Alite)、贝利特(Belite)、铝酸盐和铁素体。术语C、S、A和F在水泥化学中分别代表CaO、SiO2、Al2O3和Fe2O3。通常在粉碎熟料中添加约4%~5%的石膏(CaSO4)以调节水泥的凝结速率[21]。图3 [22]显示了水泥熟料中4种矿物化合物的结构。
《图3》
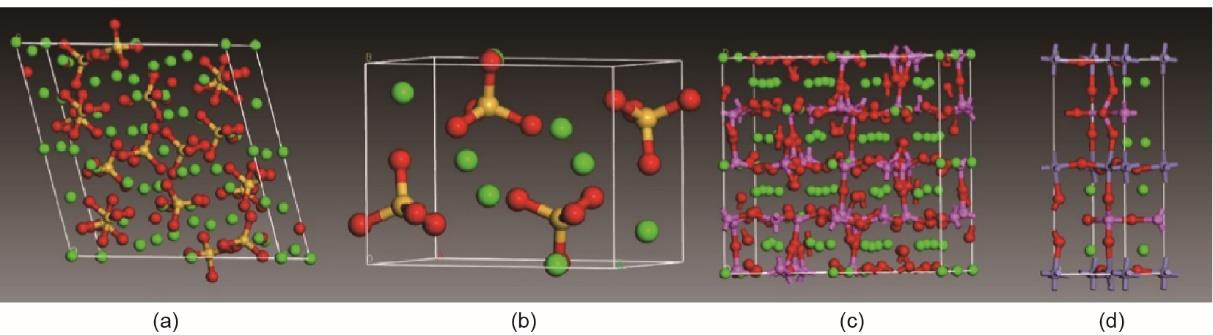
图3 水泥熟料中主要化合物的结构。(a)硅酸三钙(C3S);(b)硅酸二钙(C2S);(c)铝酸三钙(C3A);(d)铁铝酸四钙(C4AF)[
分子模拟已被用于研究水泥相的结构和性质,以提高水泥制造的效率和(或)生产所需类型的水泥。MD模拟还通过反作用力场(ReaxFF)进行,以研究C3S的表面性质和水化作用[23]。据报道,静态特性,如表面能和水吸附能,没有提供关于水化和溶解的有用数据,因此不能用来预测C3S的水化机制。在Mishra等[24]的一项研究中,对几个力场,包括COMPASS、一致价力场(CVFF)和聚合物一致性力场(PCFF)进行了参数化,以研究C3S的初始水化和内聚特性。Wu等[25]证实了COMPASS力场适用于评估硅酸盐相(C3S和C2S)的力学性能;从而通过该模拟获得了与实验数据相当的结果。此外,据报道,超级单体尺寸对水泥力学性能的影响并不显著[25]。密度泛函理论(DFT)方法也与ReaxFF一起用于研究C2S的水合[26]。此外,Tao等[27]采用从头计算来研究锰在水泥熟料中的掺杂行为。他们报道,第一原理方法提供了实验结果的基本视角。
一些研究人员还模拟了化学杂质对熟料相的结构特性和水合机制的影响。这种建模将通过优化水泥生产来降低碳排放。Huang等[28]使用DFT和MD模拟研究了杂质对C3S水化反应性的影响。原子模拟揭示了长期水化与硅酸盐水泥的电子结构之间的关系。此外,Manzano等[29]采用基于力场的方法和DFT方法研究了化学杂质(如Mg2+、Al3+和Fe3+)对主要熟料相(即阿利特和贝利特)性能的影响。据报道,有可能提高贝利特的反应性,最终有助于降低水泥生产中的能源消耗。此外,还使用MD模拟研究了C3S在硫酸钠溶液中的水化和溶解特性[30]。据报道,钠离子和硫酸根离子的存在会抑制C3S中离子的溶解,采用DFT研究水分子在C3S表面的吸附。该研究被认为是朝着更好地了解水泥水化机制迈出了一步[31]。DFT结果表明解离吸附比分子吸附更有利,分子吸附促进了水泥的溶解过程。
最近,Kumar和Mitra [32]使用INTERFACE力场(IFF)在分子水平上研究了C3A在压缩下的行为。C3A的晶胞由72个Ca、48个Al和144个O原子组成,其中,Al和O原子共价键合在褶皱环中。该模型已通过可用的实验和DFT结果进行了验证。据报道,由于键长产生的能量会影响材料在单轴和三轴压缩中的行为,而角度变形产生的能量只影响单轴压缩响应。Shahsavari等[33]也首次研究了硅酸二钙的边缘位错。据报道,γ-C2S和α-C2S晶体是贝利特中最有利于位错的多晶型,这一发现可能有助于理解水泥熟料的变形机制。
Kumar和Mitra [34]尝试在拉伸载荷下对单晶石膏进行分子动力学模拟。结合分子结构的变化以及层间和层内分离距离和层滑移讨论了单轴和三轴的应力-应变响应。据报道,石膏在拉伸载荷下表现出各向异性,这可以通过层状结构和层间水的存在来进行解释。
《3.2 水合相》
3.2 水合相
水泥作为水硬性黏合剂,能够通过水化过程与水快速反应形成新产品,这一点符合热力学和动力学规律[3]。如前所述,水化过程是最为复杂的现象之一,有关这方面的研究仍在进行中。原则上,根据方程式,硅酸盐相(大部分是C3S)的水合,主要在三个同时发生的反应[方程式(1)至方程式(3)]中进行[35]。
(1)
(2)
(3)
一般来说,水泥水化的产物可分为四个相:①水合硅酸钙(C-S-H);②氢氧化钙(CH);③钙矾石(三硫型水化硫铝酸钙,Aft);④单磺基铝酸盐(AFm)。在以下小节中,将讨论主要水合产物(C-S-H和CH)的分子模拟。
《3.2.1. 水合硅酸钙》
3.2.1. 水合硅酸钙
C-S-H占水泥浆体体积的50%~65%。许多研究人员认为C-S-H结构模型要么是胶体的,要么是层状的[36‒37];但是,仍有一个新的严格模型等待开发。事实上,这背后的原因在于水化水泥的基本组成部分的构成和结构十分复杂。然而,将描述性方法(基于实验证据)和预测模型(基于数值模拟)进行整合可以生成C-S-H的现实模型[38]。图4展示了C-S-H模型的发展历史,更多细节可以在参考文献[39‒56]中找到。C-S-H的晶胞由硅酸钙片组成,通过稳定的离子共价Ca—O和Si—O键与受限水分子连接,如图5 [57]所示。
《图4》

图4 基于实验的C-S-H形态模型和基于分子模拟的C-S-H原子结构模型。
《图5》

图5 C-S-H晶胞具有突触显示的两层富含钙和硅的区域(层内),该区域由富含水的区域(夹层)隔开[
Pellenq等[51]描述了C-S-H的分子模型,并将其指定为“现实模型”。该模型是基于C-S-H的化学特异性构建的,并使用MC模拟进行建模。使用扩展X射线吸收精细结构(EXAFS)、X射线衍射(XRD)和纳米压痕等实验技术验证了模型的Ca/Si比、密度和力学性能。Murray等[58]用正交坐标表示了C-S-H的单斜晶系。因此,使用了C-S-H的两种原子结构——晶体结构(基于Hamid模型[52])和提出的具有较暗硅酸盐链而不是连续片层的结构。还研究了C-S-H环境中中等范围Si—O和Ca—O的影响。遗憾的是,现有的基于矿物类似物的C-S-H模型与胶凝C-S-H的实验结果不一致。此外,他们无法解释原子尺度上的行为。因此,一些研究人员将DFT和MD方法结合起来,提出了一种新的模型。Manzano等[59]使用从头计算的方法来识别C-S-H的结构。
使用MD模拟详细研究了Ca/Si比率对C-S-H分子结构和力学性能的影响。使用小角中子散射(SANS)技术,C-S-H的Ca/Si比率和密度分别为1.7和2.6 g·cm-3 [60]。还有研究在不同的Ca/Si比率下研究了C-S-H的形态[61]。对两种形态——低Ca/Si比率的分支结构和高Ca/Si比率的椭圆形颗粒结构进行了区分。Masoumi等[62]研究了C-S-H的可变化学成分(具有不同的Ca/Si比率)及其纳米层和水溶液的化学反应。Kumar等[63]合成了具有从1.0到2.0变化的Ca/S比率的C-S-H,并进行了动态核极化(DNP)核磁共振(NMR)实验来表征结构的均匀性。此外,还采用原子模拟和DFT计算来测试C-S-H的结构稳定性。在水化水泥相的MM模拟中,ClayFF被证明能够很好地估计其热力学特性[64]。名为CSH-FF的经验力场是ClayFF的参数化版本,用于研究C-S-H的复杂结构[65]。
类托勃莫来石和类jnnite结构分别被广泛用于模拟C-S-H(I)和C-S-H(II)凝胶[66]。已知有不同类型的托勃莫来石矿物,包括9 Å(1 Å = 10-10 m)、11 Å和14 Å的托勃莫来石,其中,以埃为单位的长度表示特征底面间距[67‒68]。表1 [37,69‒71]给出了类矿物结构的化学结构和晶格参数。模拟中使用的托勃莫来石矿物的物理和化学性质以文献[69]中报道的实验结果为基准。使用CSH-FF模型研究了具有14 Å和11 Å结构的雪晶石,并将结果与DFT数据进行了比较[65]。Kova等[54]提出了三种基于托勃莫来石结构的模型。这些C-S-H模型随后由Kovačević等[55]进行了修改,其中研究了具有不同含水量的周期性模型集。使用ReaxFF的MD方法研究了C-S-H的有序晶体(托勃莫来石模型)和无序玻璃状凝胶[72]。另一种名为羟硅钠钙石的硅酸钙矿物也被用来模拟C-S-H的结构[70]。
《表1》
表1 水化水泥中矿物的化学结构和晶格参数
| Phase/mineral | Chemical formula | Ca/Si ratio | Lattice parameter | Density (g∙cm-3) | Ref. | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Tobermorite 9 Å | Ca5Si6O17∙5H2O | 0.83 | 11.41 | 7.42 | 11.48 | 98.36 | 97.05 | 90.23 | 2.58 | |
| Tobermorite 11 Å | Ca4Si6O15(OH)2∙5H2O | 0.67 | 6.74 | 7.451 | 22.73 | 89.63 | 90.42 | 122.22 | 2.39 | |
| Tobermorite 14 Å | Ca5Si6O16(OH)2∙7H2O | 0.83 | 6.74 | 7.425 | 27.99 | 90.00 | 90.00 | 123.25 | 2.23 | |
| Jennite | Ca9Si6O18(OH)6∙8H2O | 1.50 | 10.58 | 7.27 | 10.93 | 101.30 | 96.98 | 109.65 | 2.33 | |
| Portlandite | Ca(OH)2 | — | 3.61 | 3.61 | 4.96 | 90.18 | 89.82 | 120.02 | 2.67 | |
| Ettringite | Ca6[Al(OH)6]2(SO4)3∙26H2O | — | 11.17 | 11.17 | 21.35 | 90.00 | 90.00 | 120.00 | 1.80 | |
《3.2.2. 波特兰石》
3.2.2. 波特兰石
氢氧钙石(CH)通常被称为波特兰石,是占比第二多的水化产物,约占水化水泥的15%~25%(质量分数),具有六角形结构。它可以对许多影响耐久性的特征进行控制,包括在缓冲pH值中发挥作用,在钢筋水泥外形成亚微米的薄的保护性Fe2O3层使钢筋免受腐蚀,与硅质材料反应(火山灰反应)形成二次C-S-H,影响孔隙结构等。然而,当它被溶解时,水泥的孔隙结构可能不那么致密。由于碳酸化及酸和硫酸盐的侵蚀,它还可以作为破坏C-S-H的缓冲剂。此外,在没有波特兰石的情况下,酸或硫酸根离子将直接与C-S-H反应,从而破坏其结合能力。
由于CH在水泥基体耐久性中的重要性,使用ReaxFF [73]、DFT和MD技术检查了CH的形态。据报道,波特兰石表面上的Ca原子和O原子受到亲核和亲电力的攻击。这些研究可能有助于更好地理解CH的晶体生长。关于在纳米粒子存在下水泥的水化,Tang等[74]通过MD模拟研究了在磺化石墨烯纳米片存在下从过饱和溶液中沉淀CH。此外,使用DFT计算对CH的弹性特性进行预测[75]。
CH和硅酸盐物种(如C-S-H)生长之间的相互作用有助于理解水泥水化机制及其动力学。Galmarini等[76]使用原子模拟技术研究了硅酸盐物质在CH表面的吸附。模拟提供了有关Ca-Si复合物稳定性以及它如何影响硅酸盐水泥的生长和水化的有用信息。由于在水系统中对波特兰石晶体生长进行实验评估是困难的,MD模拟方法可以提供对原子尺度下固溶体的相互作用的基本理解[77]。据报道,在不同的吸附位点下,某些物种控制了硅酸盐的生长,反过来,这些信息将使控制水泥的可加工性成为可能。Hou等[78]研究了物质在CH纳米孔中的扩散。利用ClayFF,MD研究表明,CH表面的强水层会生成高密度和有序的组织。因此,Cl-不能长时间停留在硅酸盐弱H键的表面上。
《4、 水泥浆体的力学性能和输运性能》
4、 水泥浆体的力学性能和输运性能
《4.1 结构和力学性能》
4.1 结构和力学性能
分子模拟方法是一种强大的技术,可用于识别和量化水泥基材料的结构和力学性能。很少有研究朝这个方向进行。对未水化和水化水泥的纳米力学性能包括杨氏模量(E)、体积模量(K)、剪切模量(G)和泊松比进行了评估。原子结构的弹性常数通常使用Theodorou和Suter [79]所提出的方法进行预测。图6显示了C-S-H晶体的弹性模量(E、K和G),其中数据来自文献[80]。尽管报道的建模结果相当分散,但差异始终低于11%。
《图6》

图6 C-S-H晶体的弹性模量。Tob:托勃莫来石;线条代表拟合的线性趋势[
一般来说,C-S-H中Ca原子的存在会降低复合材料的刚度和内聚力[81]。较高的Ca/Si比率会导致C-S-H出现更多结构缺陷[82]。此外,据报道,为了提高C-S-H的拉伸强度,硅酸盐链之间的晶格间距应该变得更小[83]。Hou等[84]对C-S-H的力学性能进行了研究。使用11 Å托勃莫来石模拟不同Ca/Si比率(范围为1.3~2.0)的C-S-H结构。模拟结果表明,Ca/Si比率会对C-S-H的刚度和强度造成显著影响。
Hajilar和Shafei [68]使用类似的矿物(如托勃莫来石、羟硅钠钙石、钙矾石、云辉玢岩和水榴石),用MD方法来预测水合相的弹性特性,比较了模拟结果与文献中的实验值,发现有很大的差异。因此,为了模拟C-S-H的真实结构,Hajilar和Shafei [68]尝试通过采用微孔力学研究来考虑凝胶孔隙率。发现实验结果在通过纳米压痕技术获得的值范围内。除了MD模拟方法之外,还使用微力学模型来获得两种C-S-H的力学性能,即低密度(LD)和高密度(HD)[85]。使用微观力学技术是从多尺度角度研究C-S-H真实非晶结构的一般步骤,但尚未完全被人了解。最近,Tavakoli等[86]提出了一种组合模拟方法,包括用于确定水泥浆弹性性能的MD模拟和用于模拟微观结构的HYMOSTRUC3D方法[模拟水泥浆的三维(3D)模型]。此外,晶格模型也被用于预测水泥浆体的应力-应变响应。将所得结果与文献中可用的信息进行比较,发现结果非常一致。
Manzano等[87]试图解释理论C-S-H结果与实验发现的实际值之间的差异。他们报道说,14 Å托勃莫来石和羟硅钠钙石的弹性特性几乎是C-S-H凝胶的两倍。因此,他们表示,使用完美晶体结构的托勃莫来石和羟硅钠钙石是有问题的。此外,他们发现C-S-H的组成,主要是Ca/Si比率和水/Ca(w/Ca)比率,对水泥质C-S-H凝胶的力学性能有显著影响。尽管考虑了C-S-H中包含的孔隙率(大约30%的凝胶孔),但重新调整后的性质仍然被高估了。此外,缺陷和通过使用矿物类似物(托勃莫来石和羟硅钠钙石)带来的缺陷是由有限长度的硅酸盐链引起的。该研究得出结论,通过引入孔隙率和晶体缺陷,可以获得更好的可比较的结果。
此外,C-S-H在基本尺度上的结构和行为会在很大程度上影响水化水泥的抗拉强度和内聚力[58]。Hou等[88]在不同方向的单轴载荷下研究了胶体形式的C-S-H结构。C-S-H使用托勃莫来石建模。由于C-S-H具有层状结构,因此获得了异质的力学性能。据报道,Z方向的杨氏模量比其他方向的低50%,作者将此与沿Z方向的H键和Ca2+的存在联系起来。此外,Ca—O键对拉伸强度有显著影响。Hou等[89]还研究了具有不同Ca/Si比率并承受拉伸载荷的C-S-H结构的力学性能。结论是,高Ca/Si比会降低凝胶的拉伸强度和弹性模量。如图7 [90]所示,通过MD模拟研究了在C-S-H拉伸载荷下具有中心空隙的裂纹扩展机制[90]。模拟结果表明,中心纳米孔的存在降低了C-S-H凝胶的刚度和内聚力。Murray等[58]研究了C-S-H在单轴拉伸和压缩作用下的力学行为。他们报道说,在工程规模上,C-S-H的抗压和抗拉强度比水泥浆高一个数量级。使用MD和大规范蒙特卡洛(GCMC)方法研究了拉伸载荷与水侵蚀相结合对C-S-H凝胶产生的影响[91]。据报道,由于水侵蚀引起的水解反应导致拉伸凝胶中的内聚应力损失。研究了在外部压力作用下不同Ca/Si比率的11 Å托勃莫来石晶体的弹性行为。结果表明,20 GPa的外部压力是阈值,超过该值,弹性量发生变化。此外,随着压力的增加,Ca/Si比率为1.0的结构的弹性性能更接近Ca/Si比率为0.83的结构。
《图7》

图7 (a)C-S-H凝胶具有中央空隙,用于在单轴拉伸载荷下进行测试;(b)在不同应变率下Ca2+的位移模式。DSD:位移标准差[
使用MD模拟研究了温度变化对C-S-H力学性能的影响[93]。据报道,复合材料的体积和剪切模量随着温度的升高而降低。在的一项研究中,使用MC分子模拟研究了温度和压力对C-S-H的影响。据报道,随着温度和压力的增加,C-S-H的表面能降低。还报道了干燥和饱和状态下C-S-H凝胶的结构和性质[95]。正如宏观研究和分子模拟所观察到的,C-S-H在饱和状态下的抗压强度远大于其抗拉强度。
虽然大多数MD模拟研究都集中在C-S-H的结构和力学性能上,但Lin等[96]报道了在冲击压缩载荷下C-S-H的动力学行为。据报道,MD方法可以深入了解冲击波机制,并评估了难以通过实验测量的相关特性,如压力、比容和内能。
几个力场被用来评估水泥化合物的力学性能。具体来说,与实验结果相比,COMPASS、IFF和通用力场(UFF)的模拟结果更好[22]。力场和超胞尺寸会显著影响实验结果,因此,这些问题急需解决。
Fan和Yang [97]使用MD模拟研究了C-S-H与水分子和空隙接触的力学性能。ReaxFF被用于C-S-H建模,并沿C-S-H结构的不同方向施加单轴拉伸载荷。考虑到不同的晶格参数和晶体结构,对5种不同的原子结构(模型I到模型V)进行了建模。据报道,C-S-H的断裂力学取决于硅酸盐链,而强度在垂直于硅酸盐链的方向上显著降低。Tavakoli等[82]使用GCMC和MD方法研究了水对C-S-H弹性性能的影响。在他们的研究中,使用托勃莫来石和羟硅钠钙石结构来模拟C-S-H的结构,并使用COMPASS力场。结果表明,增加水/Ca或Ca/Si比率会降低杨氏模量。这是因为在较高的Ca/Si比率下,C-S-H的结构缺陷占主导地位,从而降低了凝胶的强度和杨氏模量。
使用原子模拟研究了C3S、C2S和C-S-H(托勃莫来石和羟硅钠钙石)的应力-应变响应[83]。还考虑了包括应变率和模拟箱尺寸在内的参数。还使用ClayFF方法研究了C-S-H(I)和C-S-H(II)的拉伸应力-应变行为[98]。Rivos Murillo等[99]研究了C-S-H在剪切下发生的变形行为,采用羟硅钠钙石进行模拟。据报道,一旦发生塑性变形,氧化钙层开始滑动,变形就开始了。这种纳米级的发现可用于防止氧化钙层的滑动,从而增强经受剪切变形的水泥浆的宏观响应。在Manzano等[100]的一项研究中,使用非平衡分子动力学(NEMD)方法报道了层间水影响下C-S-H凝胶的剪切强度。这样的研究可用于了解水泥基材料蠕变的基本机制。
使用分子静力学模拟研究了C-S-H凝胶在组合载荷下的本构行为,包括压缩或拉伸和剪切变形[101]。据报道,在纳米压痕结果下将剪切强度的压力敏感性和抗压强度与抗拉强度的不均匀性进行了定性匹配。参考文献[102]在静水压缩下研究了C-S-H的力学行为,用羟硅钠钙石结构进行模拟。据报道,压力和比容的本构关系几乎是线性的,而比内能和比容是二次方关系。Espinosa等[103]进行了一次尝试,利用MD方法提出水泥浆体的本构刚度模型。为了表示一定水化程度的水泥浆基体,所提出的模型建立在由C-S-H(类羟硅钠钙石结构)和C3S或C2S组成的二元复合体系上。据报道,所提出的复合系统表现出各向同性行为,尽管各个相具有各向异性特征。作者将两相系统的各向同性响应与各个相提供的相互作用联系起来,这种行为与水泥浆的宏观行为有关。
C-S-H的断裂特性,如表面能、断裂韧性和临界能,很难通过实验工作来进行评估。因此,MD模拟可以很好地评估这些特性[104]。据报道,水泥材料在原子尺度上的断裂特性将有助于用最少的材料来更好地设计结构。Jalilvand和Shahsavari [105]使用MD模拟研究了C-S-H的纳米级接触,包括摩擦和划痕机制。复杂系统上的这种变形机制具有挑战性,因此,MD方法对解码接触变形机制有很大帮助。
通过MD模拟研究了C3A水化产物(包括氢化石榴石、钙矾石和单硫铝酸盐)的力学行为[106],描述了这些重要产品在单轴拉伸应变下的失效机制,并基于化学键和结构损伤分析了应力-应变响应。Sarkar等[107]评估了钙矾石的变形机制。鉴于其在控制水泥早期水化速率方面发挥的重要作用,它被认为是最重要的水化产物之一。IFF用于研究钙矾石的分子变形。模拟过程包括沿钙矾石结构的所有方向施加单轴和三轴拉伸和压缩。据报道,与实验数据相比,采用的力场成功地模拟了弹性特性。此外,单轴拉伸和压缩应力-应变曲线证实了钙矾石的各向异性特性。对钙矾石和碳硫硅钙石(thaumasite)的结构和力学性能也使用DFT方法进行了预测。基于DFT结果,发现硅钙石比钙矾石更硬,这是由于前者的含水量低。
《4.2 C-S-H与水的相互作用》
4.2 C-S-H与水的相互作用
研究了水泥固体表面层间水的反应性、动力学、输运和结构特性。C-S-H凝胶中水的流动性决定了黏合相(即水泥浆)的大部分化学和物理特性(如反应性、收缩和蠕变)[108]。从MD模拟中获得的结果揭示了水分子中H原子和O原子的位置和迁移率。由于硅酸盐链中的非桥接氧原子可用作氢键,因此限制在C-S-H纳米孔中的水具有亲水性的特征[109]。特别是,由于C-S-H具有高反应性表面,水解反应发生在固液界面区[110]。因此,通过与Ca-OH和Si-OH形成相关的H键,水分子的流动性受到限制。此外,随着Ca/Si比率的增加,C-S-H中的硅酸盐骨架从有序结构转变为无定形结构。据报道,在模拟C-S-H结构的不同原子水模型中,最简单的水模型[单点电荷(SPC)] 能够提供具有良好准确性的可比结果。水化水泥性质的变化可以用水的独特性质进行定义和解释。
水分子的特性主要受相关底物结构的影响。据报道,限制在C-S-H凝胶中的水分子解离为羟基离子,从而攻击Si—O和Ca—O键[111]。在这方面,Manzano等[109]通过RAEXFF并使用MD方法研究了C-S-H凝胶中水解离的机制。据报道,在C-S-H解离后,Ca-OH和Si-OH组的形成不会影响力学性能,而会极大影响剪切强度。他们还研究了温度升高对C-S-H的层间水解离的影响[112]。限制在C-S-H纳米孔中的水与温度息息相关,其迁移率随温度而增加。MD模拟提供了在温度升高的情况下硅酸盐链断裂的信息,在温度升高时,水的水解反应降低了C-S-H中的离子共价键的产生。然而,仍需研究压力对水化水泥中水解的影响。
Yoon和Monteiro [113]在研究中发现,当温度从100 K增加到300 K时,限制在14 Å托勃莫来石层中的水的流动性会增加,但其结构保持不变。他们还研究了水在单轴张力下对石英矿物的水解弱化作用[114]。GCMC方法用于模拟水的吸附以及系统的动力学、结构和力学性能。Tang等[115]使用ClayFF研究了水分子在拉伸载荷下通过C-S-H表面的传输特性。他们构建了一个具有40 Å纳米孔的C-S-H模型,如图8 [115]所示。作者指出,所选定的力场可以很好地了解基质和液相之间的相互作用;但是,他们建议使用ReaxFF来进一步研究系统的反应性。
《图8》
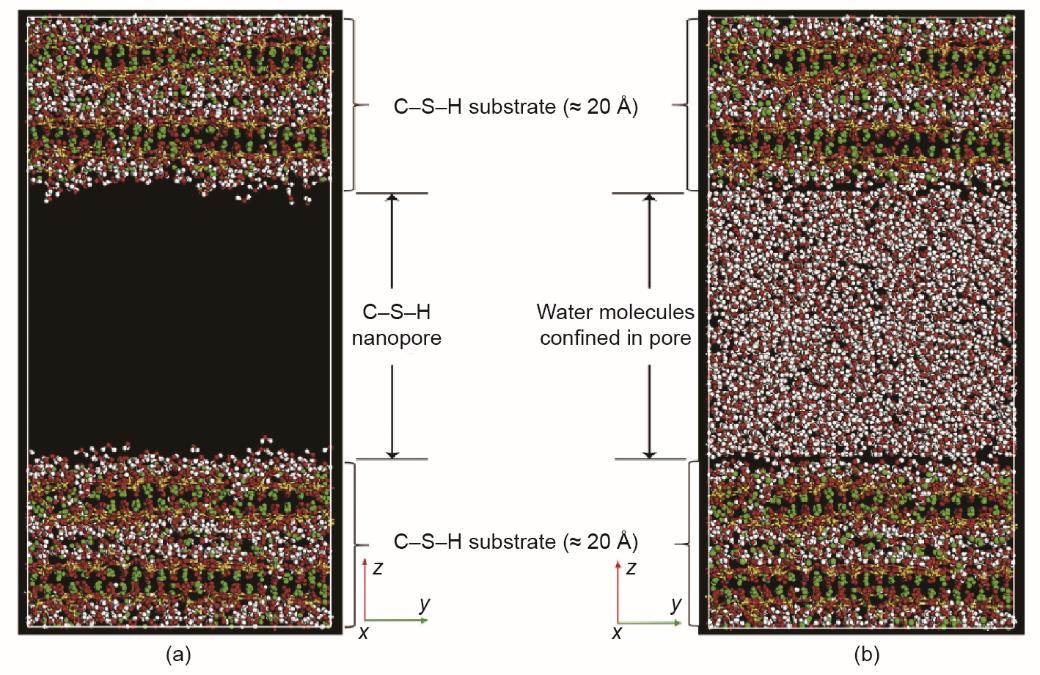
图8 (a)纳米孔C-S-H模型;(b)限制在孔隙中的水分子[
《4.3 C-S-H的传输机制》
4.3 C-S-H的传输机制
水化水泥的耐久性取决于水分子和化学物质的传输机制和界面吸附[116]。对水泥相与其他物质之间相互作用进行原子级模拟有助于更好地了解混凝土的长期耐久性。通过分子模拟研究了水分子和离子在多孔水泥介质中的迁移数据。具体来说,MD模拟方法提供了水和离子通过C-S-H凝胶孔扩散的不同分子机制[117]。使用MD模拟研究了水泥浆孔隙溶液中水和离子的动力学和结构[118]。通常,均方位移(MSD)技术用于获得自扩散系数。此外,还讨论了离子电导率在孔隙溶液中的演变[118]。
在一些研究中,将模拟结果与可用的实验数据进行了比较。Hou和Li [119]利用C-S-H的钙钛矿晶体结构对水和其他离子在其纳米孔中的传输进行了研究。在他们的模型中,MD技术是基于ClayFF使用的。作者声称,所提出的模型与35Cl NMR研究的结果非常吻合。在另一项研究[120]中,作者表明氯离子的扩散明显减少。此外,据报道,模拟研究的结果与通过NMR和准弹性中子散射(QENS)技术获得的实验数据非常吻合。他们还报道,随着与通道距离的增加,水分子的结构和动态行为会发生变化,并在距液-固界面10~15 Å处逐渐转化为本体水特性。
Hou等[121]还研究了氯离子和硫酸根离子在C-S-H夹层中的传输机制。他们首先构建了具有纳米孔通道的C-S-H模型,然后模拟水和离子的毛细流动,如图9 [121]所示。结果表明,硫酸根离子的迁移速度比氯离子慢。通过MD模拟对化学侵蚀进行模拟,并通过主动声学监测进行实验研究。据报道,C-S-H凝胶中的离子共价键在很大程度上受硫酸盐攻击的影响。Zhou等[122]通过使用ClayFF研究了氯离子和水分子通过C-S-H凝胶(建模为11 Å托勃莫来石)的传输机制。作者报道说,钙离子的存在提高了水泥对氯化物的结合能力。Hou等[123]还研究了限制在C-S-H夹层中的离子(如Ca、Mg、Al和Na离子)对内聚强度的影响。此外,Hou等[124]还讨论了氯化钠溶液在C-S-H凝胶纳米孔中的毛细管吸附。在Yang等[125]的一项研究中,对C-S-H凝胶纳米孔的尺寸对NaCl溶液传输性能的影响进行了研究。孔径范围从1 nm到3.5 nm,采用了不同的C-S-H模型。结论是,因为被氯离子和钠离子的流动所阻塞,凝胶孔的尺寸会减小。
《图9》

图9 (a)不饱和输运模型(C-S-H凝胶孔与底部溶液)和(b)NaCl + Na2SO4溶液在不同时间通过孔的毛细管吸附过程[
Yang等[126]用MD方法研究了不同Ca/Si比率下C-S-H凝胶纳米通道中硫酸盐和钠离子的吸附行为。他们使用饱和纳米孔模型来模拟在C-S-H纳米通道中溶剂化的硫酸盐和钠离子,如图10 [126]所示。结果指向两种吸附机制:SO42-与钙离子相互作用,而Na+与非桥接氧位点相连。Zhou等[127]还研究了Ca/Si比率对水分子和离子(钙离子和氯离子)传输行为的影响。据观察,随着Ca/Si比率的增加,长的硅酸盐链断裂以提供更多的氧位点以适应水和钙离子,从而吸附更多的Cl-。通过半正则MC和MD模拟以及中尺度建模探索了通过C-S-H表面的碱吸附[128]。发现钠和钾离子的吸附随着Ca/Si比率的降低而增加。
《图10》

图10 C-S-H饱和纳米孔模型,包含受限水分子、钠离子和硫酸根离子[
参考文献[129]研究了氯化钠溶液在高温下通过C-S-H凝胶扩散的情况。采用COMPASS力场,用托勃莫来石和羟硅钠钙石矿物模拟C-S-H结构。结果表明,Na+的扩散比Cl-需要更多的活化能。该模拟获得了离子的扩散系数和活化能,并与现有的实验数据进行了比较。他们还研究了NaCl溶液在0~0.05 V·Å-1的外部电场下通过C-S-H纳米孔的传输行为[130]。此类研究可以提供一些与电化学脱盐过程中离子在水泥中的扩散相关的有用信息。
除了研究C-S-H的耐久性之外,还报道了其他水泥相的研究成果。通过ClayFF并使用MD方法研究了水化水泥浆体(包括托勃莫来石、波特兰石和钙矾石)表面上的氯离子结合情况[131]。结果表明,Cl-结合能力按以下顺序递减排列:波特兰石、钙矾石和托勃莫来石。这一发现与35Cl NMR的实验结果非常吻合。Hajilar和Shafei [132]进行了水分子以及钠离子和氯离子通过单硫铝酸盐(AFm相)的传输研究。作者得出结论,内球和外球的形成控制着Na+和Cl-的扩散,其中,Cl-的迁移率比Na+的迁移率快。Huang等[133]报道了铝酸盐相中的铁的掺杂,主要是水石榴石[C3(A,F)H6],还报道了它在硫酸盐侵蚀中的作用。基于COMPASS力场的模拟结果表明,含铁相比不含铁相能够更好地对抗硫酸盐的腐蚀。使用MD模拟研究了离子在弗里德尔盐(C3A与氯离子反应形成的产物)纳米孔上的吸附和传输机制[116],观察到离子在弗里德尔盐表面的固定能力强,而水分子的动力能力大于离子。
原子模拟建模也用于研究水泥基材料与其他材料之间的相互作用。参考文献[134]报道了利用胶凝材料(C-S-H和9 Å 托勃莫来石)封装放射性锶-90的工作。此外,Bu等[135]研究了铯离子(核裂变的主要副产物)在C-S-H(托勃莫来石和羟硅钠钙石)中的吸附行为,并阐述了托勃莫来石对铯的吸附比羟硅钠钙石强。Jiang等[136]使用基于COMPASS力场的MD模拟来探索C-S-H与月桂酸(C-S-H@LA)之间的相互作用。这种复合材料可用于热能存储(TES),是相变材料(PCM)的一种应用场景。
《5、 低碳黏合剂的原子模拟》
5、 低碳黏合剂的原子模拟
如今,辅助胶凝材料(SCM)通常用于减少水泥生产对环境的影响[137]。粉煤灰和矿渣等SCM含有活性铝酸盐矿物,这些矿物与C-S-H结合形成铝硅酸钙水合物(C-A-S-H)凝胶。在包括地质聚合物在内的碱活化黏合剂中,用NaOH活化煤粉灰会产生水合铝硅酸钠(N-A-S-H)凝胶。
分子模拟也被用于研究地质聚合物,其与硬化的硅酸盐水泥具有相似的物理和化学性质。缺乏对这些材料的化学成分与宏观性能之间联系的了解,限制了它们在建筑行业的应用。因此,分子模拟可能会更好地有效了解这些黏合剂的纳米级机制和重要结构特征[138]。Sadat等[139]通过MD模拟研究了含水量和Si/Al比率变化对地质聚合物黏合剂力学性能的影响。对分布函数(PDF)用于验证模拟模型。据报道,Si/Al比率增高会加强黏合剂的刚度和拉伸强度。使用COMPASS力场研究水和氯离子在地质聚合物凝胶(A-S-H)纳米孔中的扩散[140]。在模拟系统中,A-S-H被假定为C-S-H层的形式,水分子和氯离子已植入沸石(A-S-H的类似矿物)的初始结构中。根据热力学分析,随着温度的升高,A-S-H难以吸收氯离子,因此,结构的其他部分容易发生腐蚀。
Lyngdoh等[141]提出了一种分层但无序的N-A-S-H凝胶结构,其中水分子被限制在夹层中。通过MD方法证明了离子在N-A-S-H凝胶中的输运[142]。由于N-A-S-H结构中存在非桥连氧位点,因此形成了强H键。通过使用ClayFF研究了水和硫酸盐及镁离子与N-A-S-H凝胶的相互作用。此外,利用ReaxFF [143]研究了暴露于高温下的N-A-S-H凝胶的结构。得出的结论是,在更高的温度(300~1500 K)下,水分子会分解,Al-O-Si和Si-O-Si键都会断裂,从而将凝胶转变为弱链结构。此外,由于水解反应使凝胶结构劣化,在高温和拉伸应力作用下,铝硅酸盐骨架变得脆弱。Zhang 等[144]使用MD模拟研究了N-A-S-H凝胶由于水解反应引起的拉伸行为退化。据观察,被限制的水与Si-Al骨架发生物理和化学交联。
该研究还报道了如何使用原子模拟技术研究碱活化矿渣(AAS)的产物C-A-S-H凝胶。C-A-S-H凝胶比C-S-H凝胶具有更长的链,其中,后者桥接位置的硅被氧化铝取代[145],如图11 [146]所示。ReaxFF用于研究C-A-S-H凝胶的聚合和水解反应。C-A-S-H凝胶中间层中Al原子的存在增强了黏合剂的结构和力学性能,即C-A-S-H。观察到C-A-S-H凝胶在较高的Al/Ca比率下是稳定的。此外,随着Al含量的增加,杨氏模量和抗拉强度提高。在Yang等[147]的一项研究中,用0~0.2的不同Al/Si比率对C-A-S-H凝胶进行建模。据报道,加入铝酸盐物质会产生更多的结晶顺序,Q物质的连通性(连通性因子)得到改善。此外,在较高的Al/Si比率下,由于Si-O-Al中桥接氧位点会更活跃,层间水分子会更容易解离。此外,Wan等[148]得出结论,C-A-S-H凝胶中Al和Si位点的可用性增加了结构的平均硅酸盐链长度。因此,作者建议将平均链长作为改善碱活化黏合剂性能的标准。Zhang等[149]就温度对C-A-S-H凝胶的影响进行了研究。得出的结论是,在高温下H键被破坏,因而会观察到膨胀现象。
《图11》

图11 吸收Al离子后的C-A-S-H结构[
使用MD方法研究了NaCl溶液通过C-A-S-H凝胶的传输机制[150]。采用托勃莫来石对C-S-H进行建模,然后用铝代替表面的硅构建C-A-S-H。模拟是通过具有恒定原子数、恒定体积和恒定温度(NVT)系综的ClayFF进行的。结果证实,使用纳米级研究,C-A-S-H结构中Al离子的存在限制了水分子和钠离子通过C-A-S-H的纳米孔进入凝胶。此外,Al-Si取代提供了形成氧的位点,从而导致Na—O成为稳定键。在另一项研究中,Hou和Li [151]研究了NaCl溶液和铝酸盐对C-S-H和C-A-S-H纳米孔性能的影响。据报道,在C-A-S-H中,铝酸盐硅酸盐链中氧原子贡献了更多的H键,因此受限的层状水被致密化。此外,与C-S-H 相比,C-A-S-H固定了更多的钠离子和氯离子。
Ding等[152]研究了与粒状高炉矿渣(PC-GBFS)混合的硅酸盐水泥浆体中硫酸盐侵蚀的机制。C-S-H的分子结构与11 Å托勃莫来石类似,而C-A-S-H的结构具有更多的桥连铝酸盐四面体位点。将钠离子和硫酸根离子(Na2SO4)添加到孔中以模拟硫酸盐的传输机制。在此模拟中使用了ClayFF。MD模拟结果揭示了硫酸根离子在C-A-S-H中的脱钙机制,而GBFS的添加稳定了铝硅酸盐链。在另一项研究中,通过MC方法结合MD模拟研究了硫酸根离子与C-A-S-H凝胶的相互作用[153]。C-A-S-H的结构是使用结晶C-S-H凝胶构建的,其中添加了水分子和硫酸钠(5 wt% Na2SO4)。原子模拟为硫酸盐侵蚀C-A-S-H凝胶的机制提供了一些分子级的见解。此外,还报道了其他黏合剂的MD模拟结果。以磷酸镁水泥(MPC)为例,通过其主要水化产物(即鸟粪石-K晶体)进行模拟,以探索其纳米级机制和力学行为。火山灰混凝土的性能也通过MD模拟进行了研究。具体而言,基于水-表面相互作用能理论评估了不同火山灰材料的火山灰活性指数。因此,在模拟研究的基础上,提出了一个新的方程来获得唯一的活度指数,这将简化所需火山灰材料的选择。因此,纳米级模拟可以准确预测火山灰材料的火山灰活性[154]。
《6、 结语》
6、 结语
纳米技术在水泥和混凝土科学中的应用仍处于起步阶段。近年来,通过分子模拟和水泥基材料进行建模的纳米工程研究受到了关注。本文强调了在基本尺度上对复杂水泥体系进行分子研究的重要性,并广泛回顾了在该领域开展的研究。虽然大多数研究都集中在C-S-H凝胶的力学和传输性能上,但对其组成和结构尚不清楚。因此,水泥基材料的计算建模研究仍在进行中,需要付出更多努力来开发实际的水化水泥相原子模型,主要是C-S-H模型。到目前为止,从原子模拟中获得的分子信息有助于解释在工程规模上控制混凝土行为的微观特性。此外,通过MD模拟评估的耐久性特性为水和离子通过胶凝材料的传输和扩散的纳米级机制提供了有用的见解。总之,应鼓励水泥和混凝土计算材料科学(CMS-CC)的研究和开发,以协助优化实验工作。毋庸置疑,纳米级模拟研究最终将使研究人员和工程师能够设计出新的水泥混合物或生产新一代建筑材料,以满足可持续建筑对工作性能、强度、耐久性和体积稳定性的要求,这将带来经济、技术和环境方面的效益。











 京公网安备 11010502051620号
京公网安备 11010502051620号




