
《1、 引言》
1、 引言
CO甲烷化是合成气制甲烷(CH4)的一个重要反应,其中,合成气可以从煤、生物质和有机废物的气化中获得[1‒2]。镍基催化剂已广泛用于该甲烷化反应,其中,金属Ni0为活性中心。因此,在反应之前需要进行还原预处理(催化剂活化步骤),以将新鲜催化剂中的NiO转化为金属Ni0。催化剂表面的Ni0位点的数量是催化活性的关键指标之一。这些位点与许多关键的基元反应步骤相关联,如C—O键断裂、CH氢化和CH4离解[3‒5]。因此,得到更多的金属Ni0位点是设计负载型镍催化剂的有效途径。
目前已发表的改善负载型镍催化剂性能的策略集中在合成上,例如,开发新的制备方法[6‒12],调节载体的性质[13‒15],以及添加其他掺杂元素[16]。这些方法通常用于构造分散度高且热稳定的镍纳米颗粒,从而生成更多反应位面,或增强某些基元步骤的动力学。此外,最近的研究表明,气体环境和温度对镍基催化剂的表面形态有影响。例如,发现在甲烷干重整期间引入氧化气体(如CO2和H2O),可通过从六方氮化硼纳米片载体演化的BO薄层,封装镍纳米颗粒[17]。将负载的Ni/SiO2催化剂暴露于800 ℃的CO2/H2或CH4环境中,产生了表面的Ni3C相,该相显示出电子结构的改变和弱键合线性CO吸附能力的增强[18]。然而,活化条件对催化剂性能的影响通常被忽略。根据文献,氢通常被用作还原剂,还原过程中分压、温度和持续时间共同调控还原程度和粒径分布[19‒20]。
在本工作中,我们研究了还原气氛对负载型Ni/CeO2催化剂结构的影响。用原位X射线衍射(XRD)和高分辨率透射电镜(HR-TEM)测定了金属镍纳米颗粒的晶体结构和表面形貌。用原位拉曼光谱(Raman)、准原位X射线光电子能谱(XPS)和准原位高灵敏度低能离子散射(HS-LEIS)分析了催化剂的化学状态。表面物种用原位漫反射红外傅里叶变换光谱(DRIFTS)探测。我们首次报道了Ni/CeO2的表面结构可以通过在CO甲烷化反应气体(CO/H2)中活化来进行调控,从而改善CO甲烷化反应的催化性能。
《2、 材料和方法》
2、 材料和方法
《2.1 催化剂合成》
2.1 催化剂合成
碳酸铵溶液[30%;阿拉丁试剂(上海)有限公司]作为沉淀剂,使用共沉淀法合成CeO2载体。将硝酸铈六水合物[99.95%, 0.928 g;阿拉丁试剂(上海)有限公司]溶于150 mL去离子水中。在持续搅拌的过程中,滴加碳酸铵溶液(将4.8 g碳酸铵溶于50 mL去离子水中)。将所得浆液静置12 h并抽滤。将固体在60 ℃下进一步干燥12 h,最后在500 ℃空气中煅烧4 h。
采用初湿浸渍法合成负载型Ni/CeO2催化剂。将硝酸镍六水合物[Ni(NO3)2·6H2O, 98%, 0.779 g;中国国药控股有限公司]溶于500 μL去离子水中。然后将溶液缓慢加入到制备的1.8 g CeO2粉末中,连续搅拌1 h以形成浆液。将混合物干燥(60 ℃, 12 h)并在空气中煅烧(500 ℃, 4 h)制备催化剂。NiO的标称质量负载为10 wt%。我们通过电感耦合等离子体发射光谱(ICP-OES)分析确定Ni的实际含量为8.3 wt%。
《2.2 催化剂活化》
2.2 催化剂活化
采用两种催化剂预处理条件来活化负载的Ni/CeO2催化剂。还原条件分别是:① NiCe-H:60% H2/40% N2(Air Liquide S.A.,法国),50 SCCM(标准状态下,1 SCCM = 1 mL·min-1),450 ℃,2 h;② NiCe-CO:60% H2/20% CO/20% N2(Air Liquide S.A.,法国),50 SCCM,450 °C,2 h。
《2.3 催化剂表征》
2.3 催化剂表征
使用透射电子显微镜(TEM,在200 kV的电压下操作;FEI Tecnai G2 F20 S-Twin, FEI Company,美国)表征催化剂的形貌。能量色散X射线光谱(EDS)用于元素分析。
原位拉曼光谱在Horiba LabRam HR光谱仪(Horiba, Ltd.,日本)上收集,该光谱仪配备波长为514 nm的可见激光激发(光源:He-Cd)、50倍长的工作距离物镜(Olympus BX-30-LWD, Olympus Corp.,日本)、单级单色仪和电荷耦合器件(CCD)检测器(Horiba CCD-3000V)。将催化剂活化2 h并在450 ℃下CO甲烷化1 h,每15 min收集一次光谱。
原位XRD分析在具有在40 kV和40 mA下操作的CuKα射线源(λ = 0.154 nm)的Bruker D8 ADVANCE X射线粉末衍射仪(Bruker Corp.,美国)上进行。催化剂在450 ℃ Ar中处理并在不同条件下活化。然后从20°~80°收集XRD图案,分辨率为0.02°。
通过配备有单色AlK辐射源[1486.6 eV (1 eV = 1.602176×10-19 J),通过能量为20.0 eV]的Thermo ESCALAB 250Xi光谱仪(Thermo Fisher Scientific,美国)收集准原位XPS光谱。预处理室用于催化剂活化。然后将预处理室抽空,并将样品在真空中快速转移到分析室中而不与空气接触。
用配备有双环面分析仪的Qtac 100 HS-LEIS光谱仪(ION-TOF,德国)分析表面元素分布。样品通过在2000 psi (1 psi = 6.894757×103 Pa)的坩埚内被压缩成Al2O3粉末来制备并用于分析。将沉淀装入预处理室,在10% O2/Ar环境中在450 ℃的温度下脱水1 h,并在两种条件下活化:① 1∶1 H2/Ar和② 1∶3 CO:H2在350 ℃下保持2 h。然后,将沉淀物转移到分析室以避免空气接触。使用4 keV的He+作为探针离子源,使用0.5 keV的Ar+进行溅射。
H2程序升温还原(TPR)在连接到在线热导检测器(TCD)的Autosorb-TP-5080-B(天津市先权仪器有限公司)上进行。温度以10 ℃·min-1的速度升至800 ℃,与10% H2/Ar在30 sccm的流速下保持30 min。
通过使用Micromeritics AutoChem II测量化学吸附的氢分子的量来估计金属Ni的表面积。催化剂在450 ℃下在Ar中预处理并在不同条件下活化。然后,用Ar吹扫催化剂以除去物理吸附的H2,冷却至室温,进行H2化学吸附脉冲。
原位DRIFTS实验在配备有漫反射池(Harrick Praying Mantis, Harrick Scientific Products Inc.,美国)的PerkinElmer Frontier傅里叶变换红外(FT-IR, PerkinElmer, Inc.,美国)光谱仪上进行。将约50 mg催化剂加载到反应池中。首先在450 ℃的不同处理条件下活化催化剂。随后,将温度控制在300 ℃,并将气流切换至CO甲烷化反应气体1 h。在反应期间每分钟收集原位DRIFTS光谱。
《2.4 稳态活性测试》
2.4 稳态活性测试
在固定床微反应器中测试CO甲烷化性能:将100 mg催化剂粉末置于石英管中,在不同气氛下活化后,切换至50 sccm的20% CO/60% H2/20% N2,在200~450 ℃的温度下进行CO甲烷化反应。所有活性测试均在30 000 mL·g-1·h-1或1 500 000 mL·g-1·h-1的气体空速(GHSV)下进行。采用在线气相色谱法(使用上海锐敏GC 2060气相色谱仪)分析气体产物。使用5A分子筛和Porapak T柱(Sigma-Aldrich Inc.,美国)进行气体分离。通过TCD检测H2;碳氢化合物、CO2和CO由配备甲烷转化炉的火焰离子化检测器(FID)检测。
《3、 结果和讨论》
3、 结果和讨论
将负载的Ni/CeO2催化剂在450 ℃下还原2 h,并评估CO甲烷化反应的活性。选择了两种不同的活化气氛:60% H2/40% N2和20% CO/60% H2/20% N2;还原催化剂分别表示为NiCe-H和NiCe-CO。NiCe-H催化剂在200 ℃和30 000 mL·g-1·h-1的条件下,CH4的产率和CO的产率分别为146.3 mmol·g-1·h-1和31.8 mmol·g-1·h-1 [图1(a)和附录A中的表S1]。相比之下,NiCe-CO对CO甲烷化反应更为活跃,相同反应条件下,CH4的产率为163.0 mmol·g-1·h-1。在较高的GHSV下,催化活性的改善变得更加明显。与NiCe-H (95.8 mmol·g-1·h-1)相比,NiCe-CO (121.8 mmol·g-1·h-1)的CH4产率提高了约30%。
《图1》

图1 (a)不同预处理方法的负载Ni/CeO2催化剂上CH4和CO的产率;(b)不同预处理方法的负载Ni/CeO2催化剂上的24 h稳定性测试[反应条件:P = 1.0 atm, T = 200 ℃, GHSV = 30 000 mL∙g-1∙h-1, n(H2):n(CO) = 3:1]。
还原是激活催化剂的必要步骤,未还原的催化剂对CO甲烷化反应的活性可以忽略不计。两种活化催化剂在24 h内表现出非常稳定的性能[图1(b)],这意味着不同的活化条件可能导致催化剂结构发生某种不可逆的变化。我们还评估了活化条件对Ni/CeO2催化剂上CO甲烷化反应的影响,该催化剂具有较高的标称NiO负载量(50wt%)。由反应物活化的催化剂仍显示出更好的性能(附录A中的表S2)。
我们分析了负载Ni/CeO2催化剂在不同预处理后的形貌。其表面积和孔隙率似乎较为接近(附录A中的图S1、图S2、表S3),表明上述结构变化最有可能与负载的金属镍纳米颗粒相关。NiCe-H催化剂的HR-TEM图像显示具有Ni (111)位面(0.21 nm)晶格条纹的较大的纳米颗粒(图2)。根据EDS图谱,金属镍纳米颗粒的平均尺寸约为21.6 nm。有趣的是,虽然从EDS图谱中获得的NiCe-CO的表观粒径(18.7 nm)与氢处理的催化剂较为接近,但这些Ni0颗粒呈多晶状态,具有纳米尺寸的颗粒(平均直径为7.9 nm)。用原位XRD进一步探测活化前后Ni/CeO2的体结构,发现CeO2相位于28.3°、32.7°、46.9°和55.8° [图3(a)]。根据CeO2 (111)衍射峰,使用Scherrer方程确定CeO2的晶粒尺寸为11.8 nm。对于NiCe-H (12.5 nm)和NiCe-CO (11.8 nm),它几乎保持不变。对于新催化剂,可以检测到NiO相,其在43.1°处具有微小的NiO (200)峰,对应于20.9 nm的晶粒尺寸(附录A中的图S3)。NiCe-H在44.0°处有一个衍射峰,这是Ni (111)相的特征峰。Ni的晶粒尺寸为22.6 nm。然而,NiCe-CO很难检测到与金属镍相关的衍射峰,这与HR-TEM结果一致。
《图2》
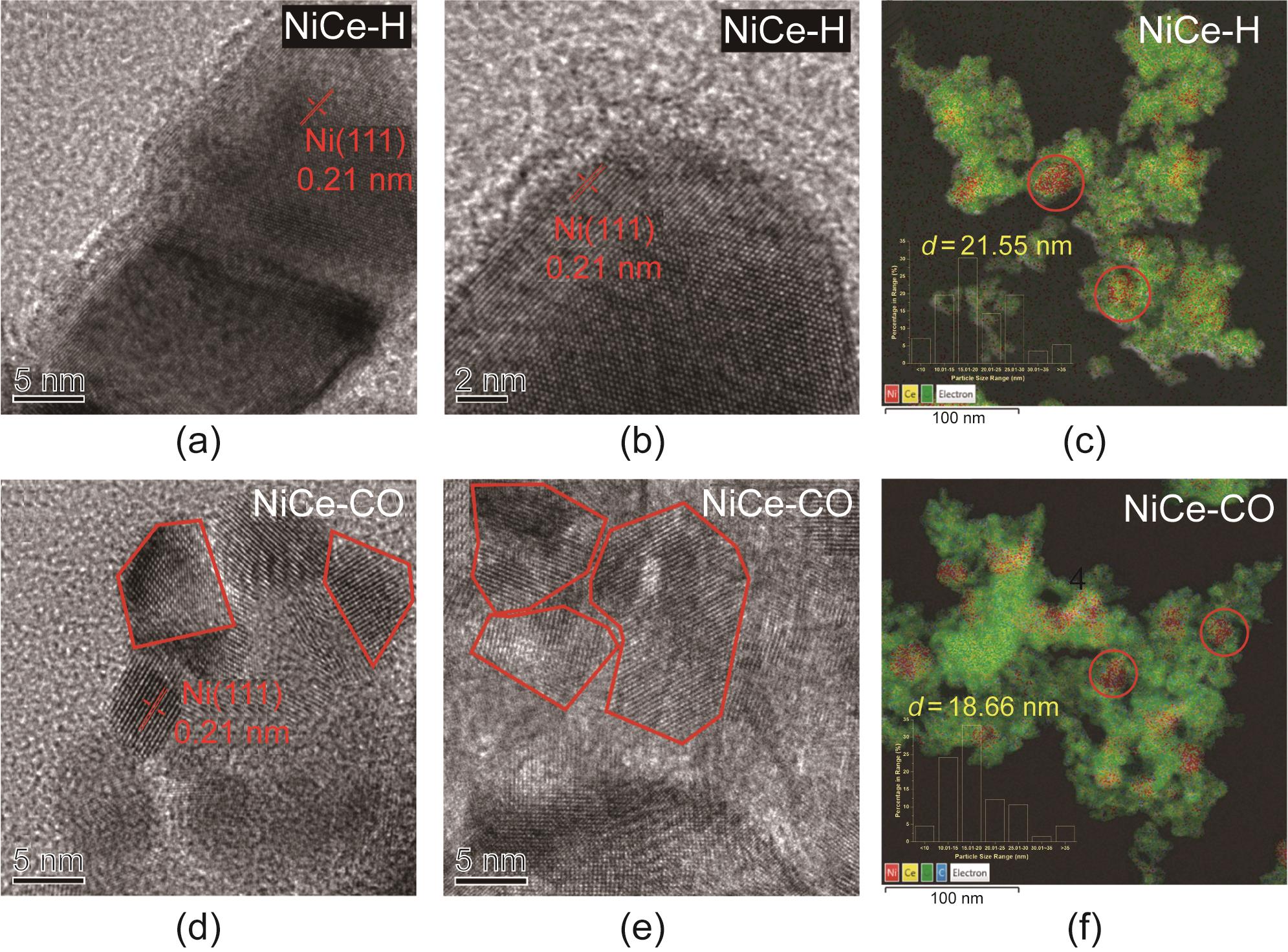
图2 NiCe-H [(a)~(c)]和NiCe-CO [(d)~(f)]的HR-TEM图像和EDS面扫图像。
《图3》
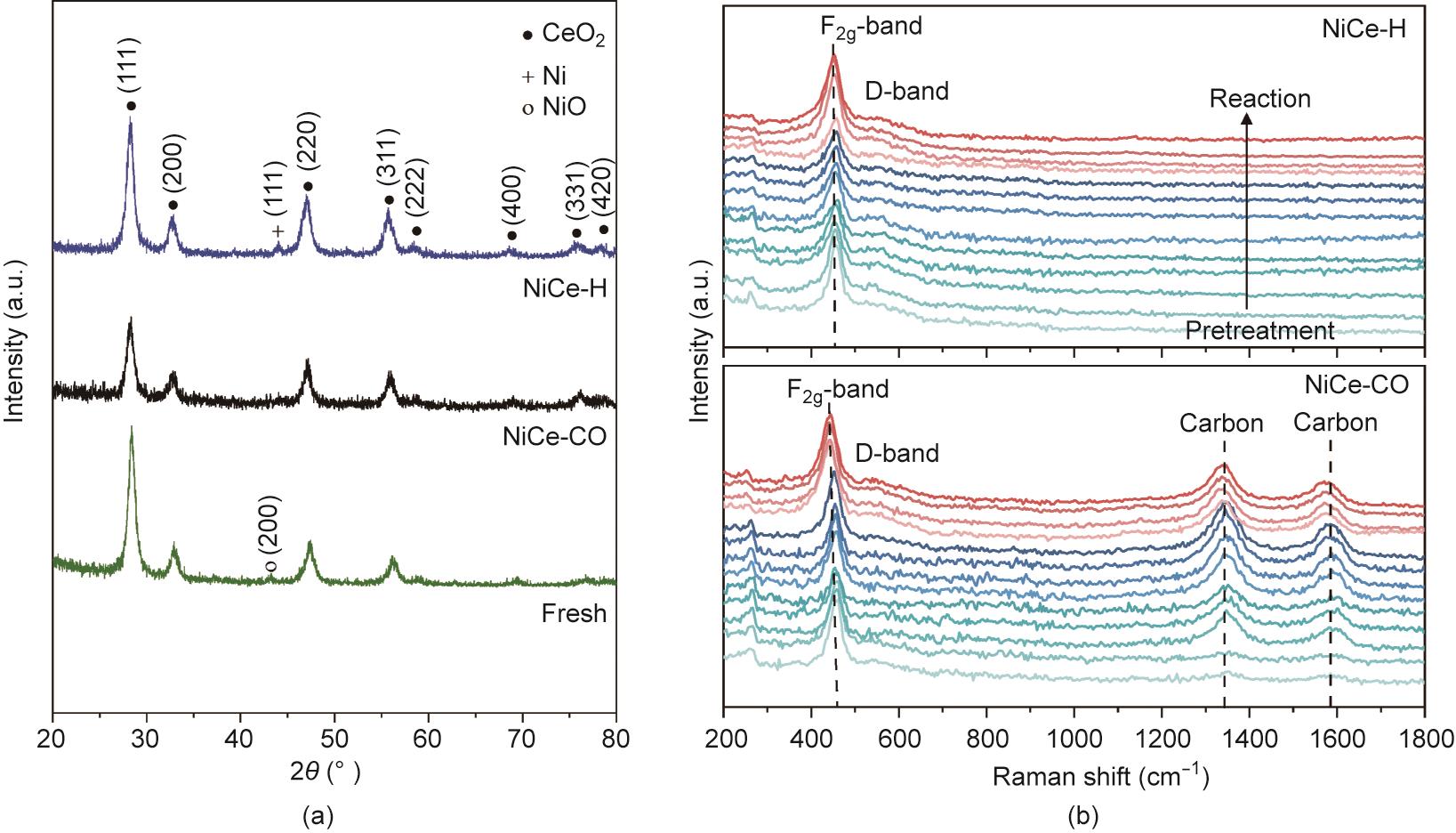
图3 (a)负载Ni/CeO2催化剂原位XRD图像;(b)NiCe-H和NiCe-CO活化过程和反应过程的时间分辨的原位拉曼光谱图像。
通过时间分辨原位拉曼光谱还跟踪了催化剂在活化和反应期间的结构演变。尖峰位于约460 cm-1处为F2g band,而位于约590 cm-1的宽峰为缺陷诱导模式(D band)[图3(b)] [21‒23]。F2g band的位置在某种程度上与CeO2中的缺陷位(即氧空位)数量有关。该位置还可能受到晶格应变和颗粒尺寸的影响[16,23‒24]。Ni/CeO2的活化产生了氧空位,更强的D band和发生红移的F2g band证明了这一点。在CO甲烷化反应期间,这些氧缺陷似乎相当稳定,D band的强度几乎保持不变。
积碳表现在1357 cm-1处的D band和1585 cm-1处的G band。这些物质在活化过程中通过CO和H2的混合物形成[25]。已有大量文献记录了含CO气氛中碳物种的形成。例如,通过5% Co/He活化Pt/Co/Al2O3,产生了用于费-托合成的微孔碳壳[26]。有趣的是,氢活化的催化剂(NiCe-H)处于CO甲烷化反应条件下时,即使气体环境和温度与NiCe-CO的活化方法相同,都不会产生任何碳物种。此外,积碳的能带强度在反应阶段保持恒定。我们推测,碳物种的形成仅伴随着在CO/H2环境中NiO向金属Ni纳米颗粒的相变。这样的特征可以解释如上所示的CO甲烷化期间优异的长期稳定性(无积碳形成)。
用准原位XPS进一步研究了催化剂表面区域的化学状态。新鲜Ni/CeO2在855.0 eV (85%)下表现出主要由Ni2+物种贡献的Ni 2p光谱,在853.2 eV (15%)下有少量的Ni0物种[图4(a)]。新鲜催化剂中的Ni0物种可能是由于具有氧缺陷的CeO2-和NiO之间的电子相互作用。活化后,金属Ni0物种的含量显著增加,NiCe-H和NiCe-CO分别达到60%和66%。Ce 3d信号的XPS光谱揭示了Ce4+位点[Ce3d94f0O2p6(μ‴和ν‴)、Ce3d94f1O2p5(μ″和ν″)、Ce3d94f2O2p4(μ和ν)]以及Ce3+位点[Ce3d94f2O2p5(μ0和ν0)和Ce3d94f1O2p6(μ′和ν′)] [27]。与原位拉曼结果一致,预处理分别将NiCe-H和NiCe-CO的氧空位数提高到43%和46%。然而,活性Ni/CeO2催化剂的相应O 1 s光谱显示无差异(附录A中的图S4)。总的来说,我们的XPS结果表明,两种活化方法都能还原催化剂,还原程度和表面元素分布相似(附录A中的表S4)。我们还对Ni/CeO2催化剂进行了H2-TPR分析,两种活性催化剂显示了可比较的H2吸收,这与XPS结果保持一致(附录A中的图S5)。
《图4》
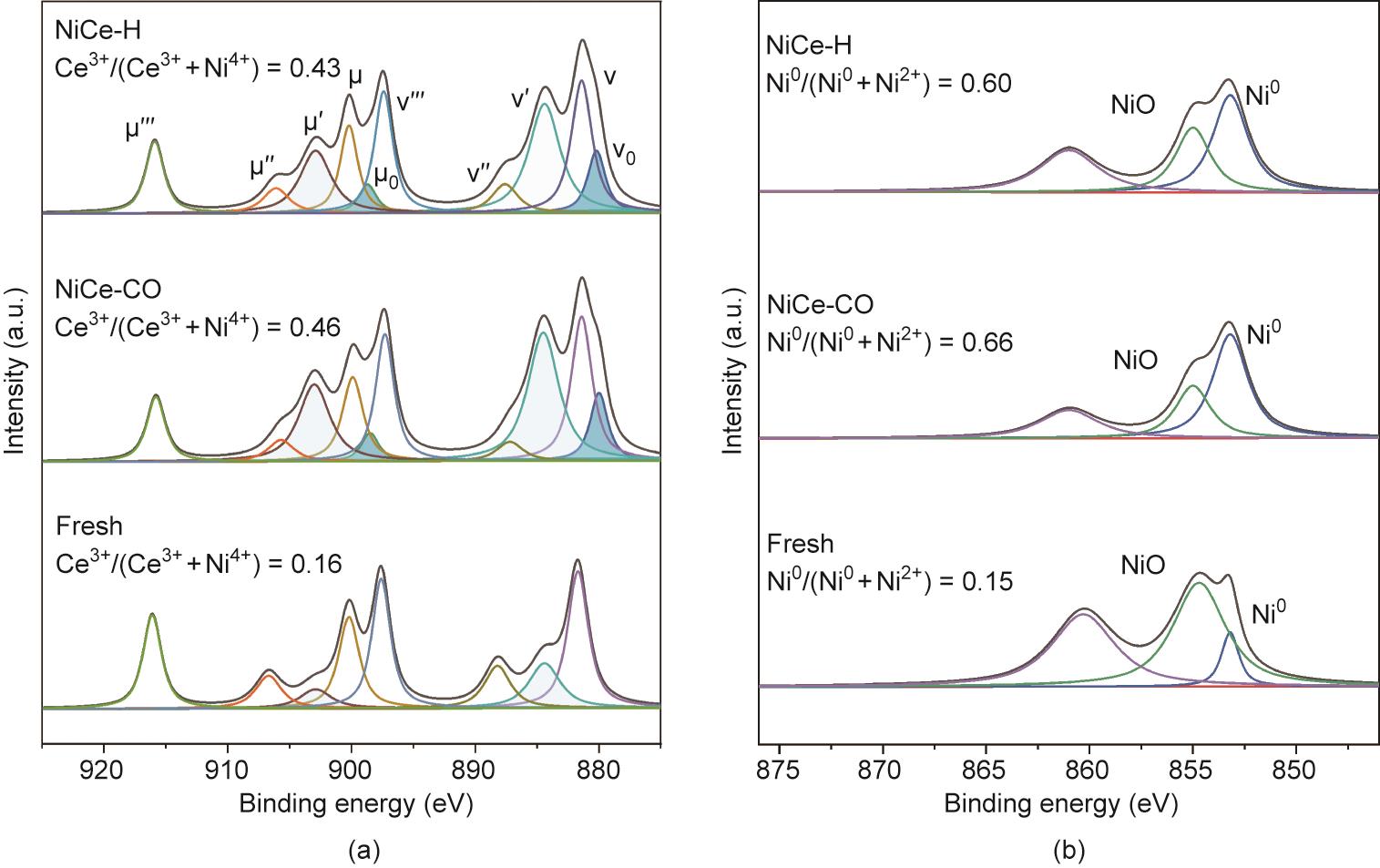
图4 不同预处理方法的负载Ni/CeO2催化剂上准原位XPS图像。(a)Ce 3d区域;(b)Ni 2p区域。
Ni和CeO2的表面氧化态不受活化过程中添加CO的影响;因此,我们假设NiCe-CO优异的CO甲烷化性能与金属镍纳米颗粒的多晶性质有关。多晶纳米颗粒含有晶界,通常具有优异的催化活性[28‒30]。通过原位DRIFTS分析探讨了催化剂的表面条件。催化剂被活化后,用Ar吹扫,随后切换为反应气体。在300 ℃下引入CO甲烷化反应物后,我们通过CO与表面羟基的相互作用观察到表面甲酸盐物种,并通过CO2副产物的再吸附观察到表面碳酸盐物种(附录A中的图S6)[31]。线性吸附的*CO物种逐渐累积,从约2140 cm-1到2050 cm-1略有红移[图5(a)和(b)] [31]。在CO/H2混合物活化期间也会形成*CO物种,但没有明显的峰移[图5(c)]。在反应的初始阶段,*CO信号的红移很可能与Ar吹扫期间产生的轻度氧化镍表面的还原有关。NiCe-CO的*CO物种的红外(IR)峰值强度似乎远高于NiCe-H的峰值强度[图5(d)],这表明有更多的CO吸附位点(即暴露的Ni0位点)[32]。与NiCe-H (0.85 m3·g-1)相比,NiCe-CO (1.24 m3·g-1)在H2化学吸附过程中的H2吸附量更大,这进一步证明了NiCe-CO有更多的CO吸附位点(附录A中的图S7)。
《图5》
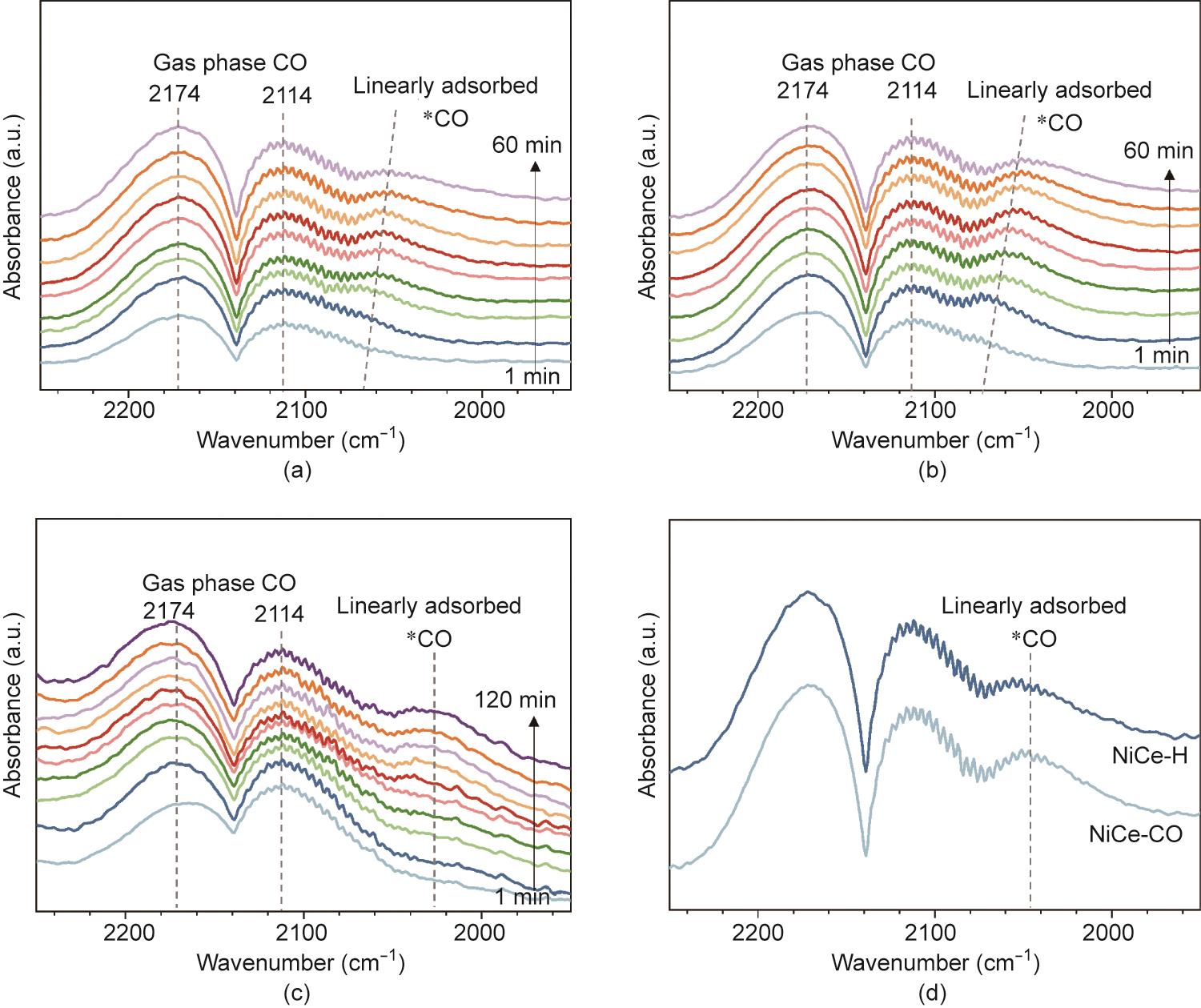
图5 NiCe-H(a)和NiCe-CO(b)在300 ℃反应条件下的原位DRIFTS图像;(c)Ni/CeO2催化剂在CO/H2活化过程中的原位DRIFTS图像;(d)NiCe-H和NiCe-CO在300 ℃反应条件下反应60 min后的IR对比图像。
这些数据表明活性Ni/CeO2催化剂的构效关系。在活化过程中,NiO被还原为金属Ni,这为C—O键断裂和CH氢化提供了活性位点[3‒4]。在CO/H2活化Ni/CeO2的过程中,碳物种沉积在催化剂表面上。同时,Ni在NiCe-CO样品中的颗粒呈现出多晶形态,晶粒尺寸为纳米级。目前的研究结果表明,在CO/H2中活化后,Ni0位面的分布存在差异,这一点尚待报道。但是,之前有许多研究描述了反应期间气体分子/表面吸附质与催化剂表面之间的相互作用[33‒36]。我们假设此实验中的情况是由于积碳在镍纳米颗粒的不同位置引入了各向异性晶格应变[37‒39]。这些碳物种可能覆盖一部分金属镍纳米颗粒,但是从准原位HS-LEIS对NiCe-H和NiCe-CO的最外层表面的Ni原子分析来看,催化剂仍含有大量的Ni0位点(附录A中的图S8)。除非形成聚合碳或致密有序石墨层,否则这些碳物种通常是多孔的,对催化活性的影响较小[40]。反应物活化催化剂的多晶性质导致许多晶界显示出更大的CO吸附容量和更高的表观甲烷化活性。然而,镍晶界处CO甲烷化反应的本征反应动力学尚未被阐明,需要进一步详细的动力学和理论分析。
《4、 结论》
4、 结论
总之,我们发现不同的预处理条件会影响负载型Ni/CeO2催化剂的表面结构。与使用氢气的传统活化方法不同,在CO/H2混合气体中对催化剂进行预处理,会产生多晶镍纳米颗粒,这些纳米颗粒具有丰富的晶界和更大的CO吸附容量,有利于CO甲烷化。多晶镍纳米颗粒的形成可能与表面上的碳沉积有关。本研究提供了一种通过调控活化条件来优化催化剂结构的新策略。











 京公网安备 11010502051620号
京公网安备 11010502051620号




